Context-Dependent Role of IKKβ in Cancer

 , , , and
, , , and
Abstract
1. Introduction
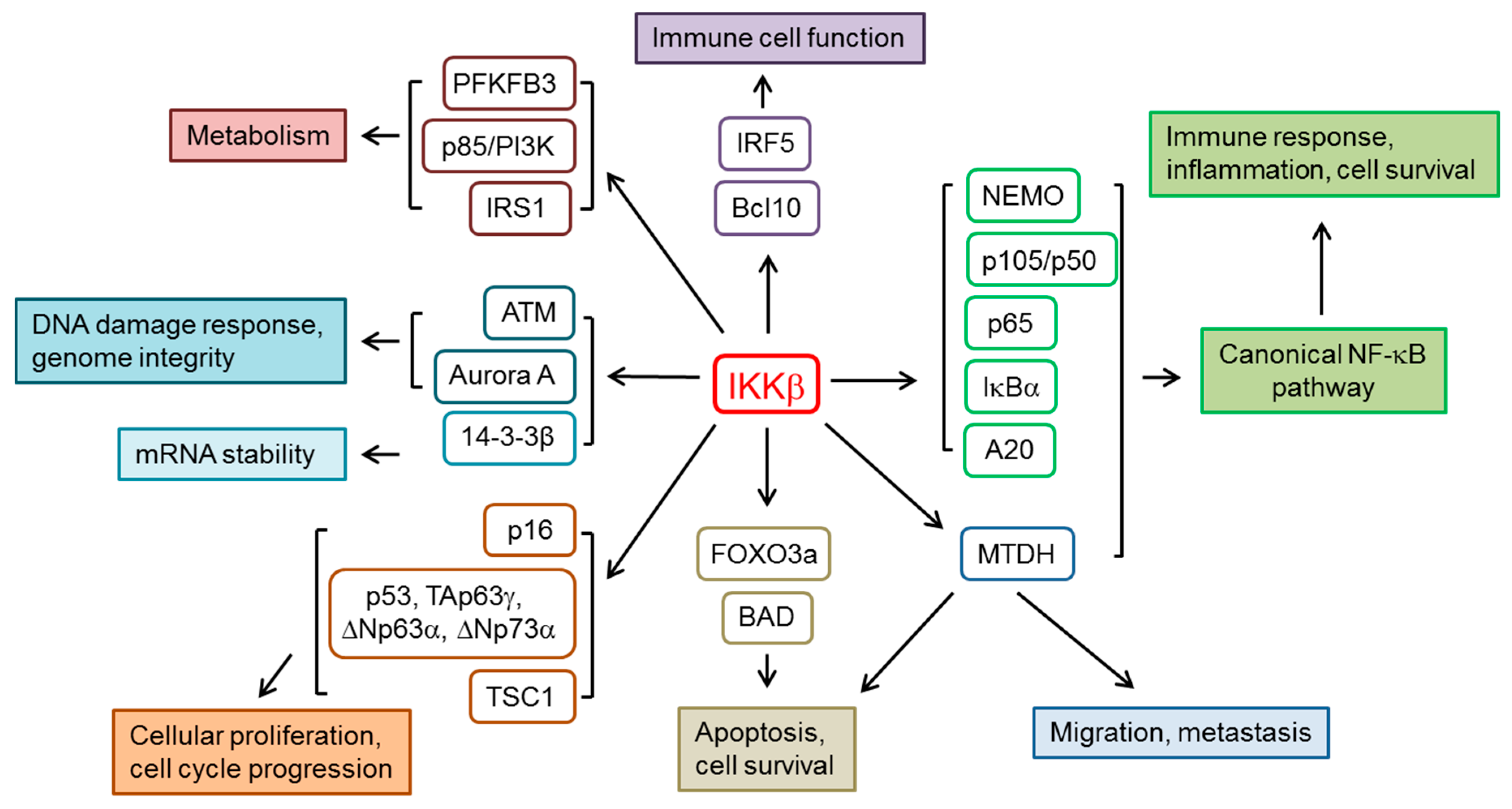
2. IKKβ and the Regulation of NF-κB Pathway
3. Other IKKβ Substrates and Cellular Functions
3.1. Cellular Proliferation and Cell Cycle Progression
3.2. Immune Cell Function
3.3. Metabolism
3.4. DNA Damage, Genome Integrity and mRNA Stability
3.5. Apoptosis, Cell Survival and Cell Migration
4. Mouse Models for the Study of IKKβ Role in Cancer
4.1. Lung Cancer
4.2. Melanoma
4.3. Pancreatic Cancer
4.4. Liver Cancer
4.5. Intestinal Cancer
4.6. Non-Melanoma Skin Cancer, Oral and Esophageal Cancer
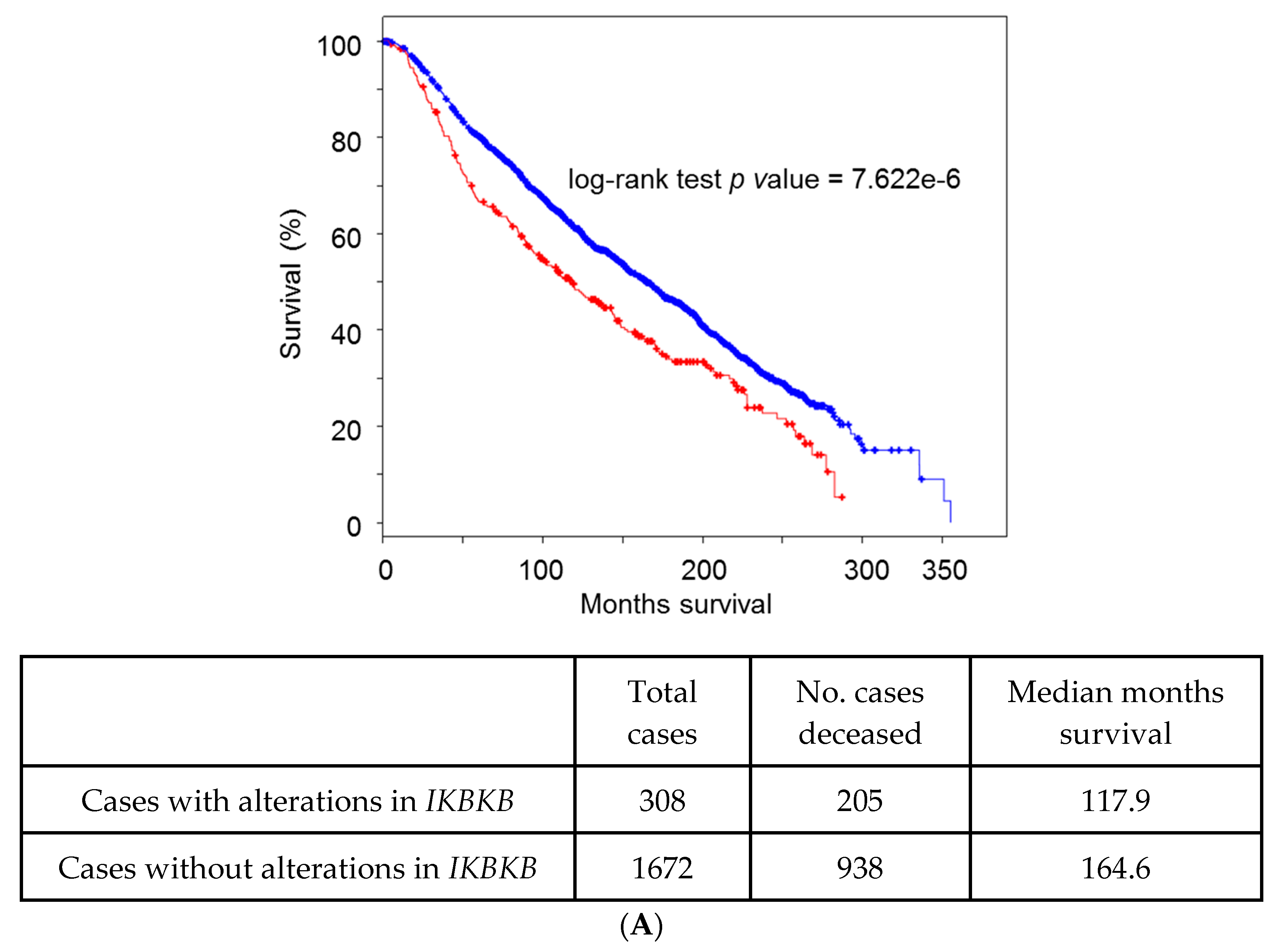
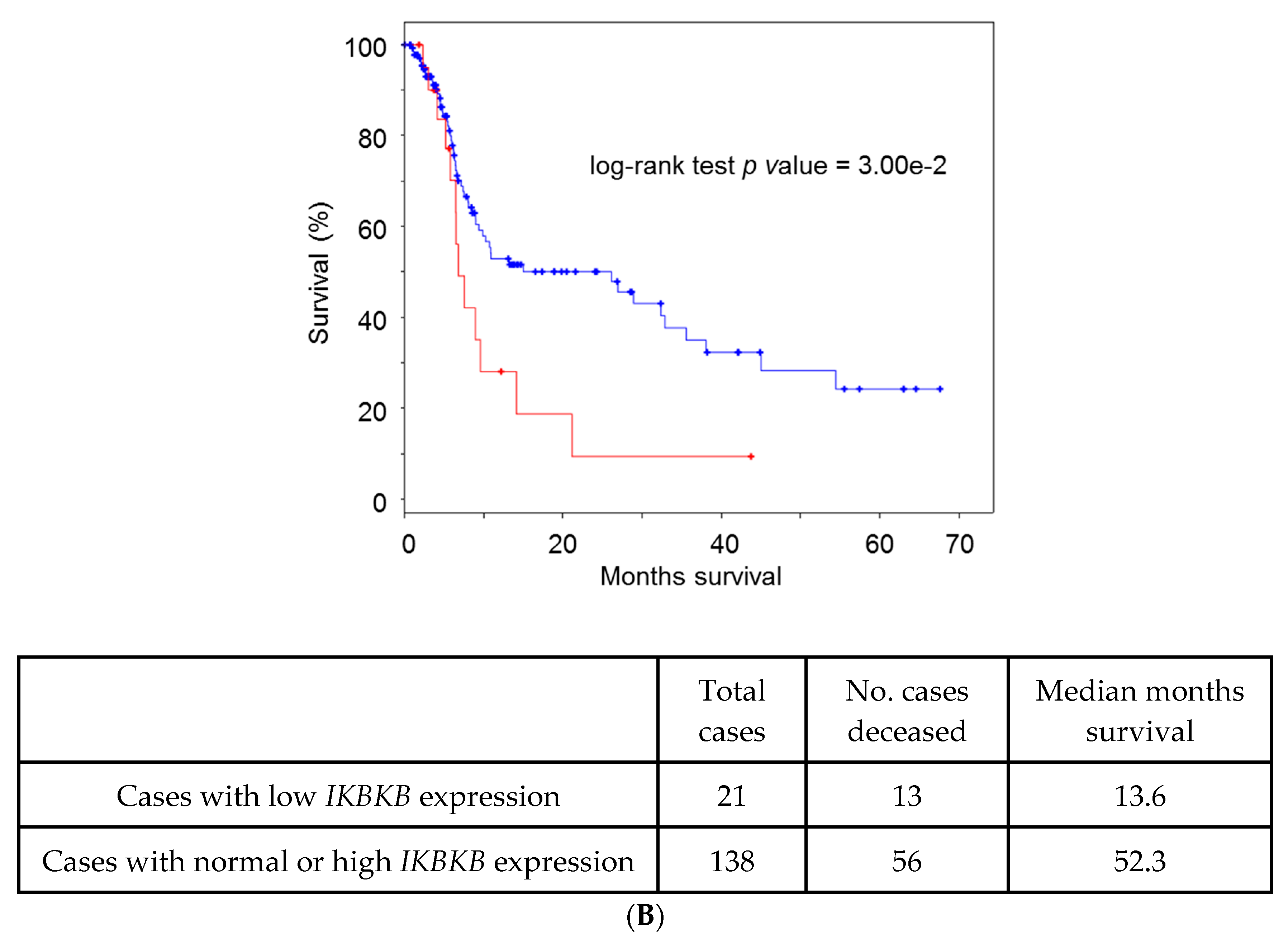
5. Lessons from Human Tumoral Samples
Acknowledgments
Conflicts of Interest
References
- Forbes, S.A.; Bhamra, G.; Bamford, S.; Dawson, E.; Kok, C.; Clements, J.; Menzies, A.; Teague, J.W.; Futreal, P.A.; Stratton, M.R. The catalogue of somatic mutations in cancer (COSMIC). In Current Protocols in Human Genetics; Haines, J.L., Korf, B.R., Morton, C.C., Seidman, C.E., Smith, D.R., Eds.; Johns Wiley and Sons: Hoboken, NJ, USA, 2008; Chapter 10, Unit 10; ISBN 9780471142904. [Google Scholar]
- Catalogue of Somatic Mutations in Cancer (COSMIC). Available online: http://cancer.sanger.ac.uk/cosmic (accessed on 18 October 2017).
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD4(+) T cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; Scoggins, R.M.; Han, W.; Polosukhin, V.V.; Connelly, L.; Yull, F.E.; Fingleton, B.; Blackwell, T.S. Epithelial NF-κB activation promotes urethane-induced lung carcinogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18514–18519. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Arslan, S.C.; Scheidereit, C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol. Rev. 2012, 246, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Krappmann, D.; Wulczyn, F.G.; Scheidereit, C. NF-κB p105 is a target of IκB kinases and controls signal induction of BCL-3–p50 complexes. EMBO J. 1999, 18, 4766. [Google Scholar] [CrossRef] [PubMed]
- Waterfield, M.; Jin, W.; Reiley, W.; Zhang, M.; Sun, S.C. IκB kinase is an essential component of the TPL2 signaling pathway. Mol. Cell. Biol. 2004, 24, 6040–6048. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, E.; Guan, K.; Wang, C.Y. IKKβ plays an essential role in the phosphorylation of RELA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 2003, 170, 5630–5635. [Google Scholar] [CrossRef] [PubMed]
- Hutti, J.E.; Turk, B.E.; Asara, J.M.; Ma, A.; Cantley, L.C.; Abbott, D.W. IκB kinase β phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Mol. Cell. Biol. 2007, 27, 7451–7461. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, non-catalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Palkowitsch, L.; Leidner, J.; Ghosh, S.; Marienfeld, R.B. Phosphorylation of Serine 68 in the IκB kinase (IKK)-binding domain of NEMO interferes with the structure of the IKK complex and tumor necrosis factor-α-induced NF-κB activity. J. Biol. Chem. 2008, 283, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Lopez, T.; Israel, A.; Weil, R. Negative feedback loop in t cell activation through IκB kinase-induced phosphorylation and degradation of BCL10. Proc. Natl. Acad. Sci. USA 2007, 104, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Padre, R.C.; De Mendoza, T.H.; Bottero, V.; Tergaonkar, V.B.; Verma, I.M. Phosphorylation of p53 by IκB kinase 2 promotes its degradation by β-TRCP. Proc. Natl. Acad. Sci. USA 2009, 106, 2629–2634. [Google Scholar] [CrossRef] [PubMed]
- Ak, P.; Levine, A.J. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.M.; Zhang, Y.; Liao, W.; Zeng, S.X.; Su, X.; Flores, E.R.; Lu, H. IκB kinase β (IKKβ) inhibits p63 isoform γ (TAP63γ) transcriptional activity. J. Biol. Chem. 2013, 288, 18184–18193. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Chang, X.; Sen, T.; Ravi, R.; Bedi, A.; Sidransky, D. Regulation of p53 family member isoform Δp63α by the nuclear factor-κB targeting kinase IκB kinase β. Cancer Res. 2010, 70, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Scalise, M.; Gheit, T.; Hussain, I.; Yue, J.; Carreira, C.; Collino, A.; Indiveri, C.; Gissmann, L.; Sylla, B.S.; et al. IκB kinase β promotes cell survival by antagonizing p53 functions through Δp73α phosphorylation and stabilization. Mol. Cell. Biol. 2011, 31, 2210–2226. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yuan, C.; Weghorst, C.M.; Li, J. Ikkβ specifically binds to p16 and phosphorylates Ser8 of p16. Biochem. Biophys. Res. Commun. 2010, 393, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK β suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Supprian, M.; Courtois, G.; Tian, J.; Coyle, A.J.; Israël, A.; Rajewsky, K.; Pasparakis, M. Mature T cells depend on signaling through the IKK complex. Immunity 2003, 19, 377–389. [Google Scholar] [CrossRef]
- Heuser, C.; Gotot, J.; Piotrowski, E.C.; Philipp, M.S.; Courreges, C.J.F.; Otte, M.S.; Guo, L.; Schmid-Burgk, J.L.; Hornung, V.; Heine, A.; et al. Prolonged IKKβ inhibition improves ongoing CTL antitumor responses by incapacitating regulatory T cells. Cell Rep. 2017, 21, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pelaez, M.; Lamont, D.J.; Peggie, M.; Shpiro, N.; Gray, N.S.; Cohen, P. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 17432–17437. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chen, X.; Chen, Z.J. IKKβ is an IRF5 kinase that instigates inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 17438–17443. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor κ B kinase complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef] [PubMed]
- Comb, W.C.; Hutti, J.E.; Cogswell, P.; Cantley, L.C.; Baldwin, A.S. P85alpha sh2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and AKT in response to cellular starvation. Mol. Cell. 2012, 45, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Lowman, X.H.; Pan, M.; Tran, T.Q.; Warmoes, M.O.; Ishak Gabra, M.B.; Yang, Y.; Locasale, J.W.; Kong, M. IKKβ promotes metabolic adaptation to glutamine deprivation via phosphorylation and inhibition of PFKFB3. Genes Dev. 2016, 30, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Margalef, P.; Bigas, A. Non-conventional functions for NF-κB members: The dark side of NF-κB. Oncogene 2015, 34, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hikiba, Y.; Nakagawa, H.; Hirata, Y.; Hayakawa, Y.; Kinoshita, H.; Nakata, W.; Sakitani, K.; Takahashi, R.; Akanuma, M.; et al. Promotion of DNA repair by nuclear IKK[β] phosphorylation of ATM in response to genotoxic stimuli. Oncogene 2013, 32, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Irelan, J.T.; Murphy, T.J.; DeJesus, P.D.; Teo, H.; Xu, D.; Gomez-Ferreria, M.A.; Zhou, Y.; Miraglia, L.J.; Rines, D.R.; Verma, I.M.; et al. A role for IκB kinase 2 in bipolar spindle assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 16940–16945. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shin, E.M.; Lee, S.; Mathavan, S.; Koh, H.; Osato, M.; Choi, H.; Tergaonkar, V.; Korzh, V. IKK2 regulates cytokinesis during vertebrate development. Sci. Rep. 2017, 7, 8094. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Garcia-Vallejo, J.J.; van Het Hof, B.; van Dijk, W. Convergent actions of IκB kinase β and protein kinase Cδ modulate mRNA stability through phosphorylation of 14–3-3β complexed with tristetraprolin. Mol. Cell. Biol. 2005, 25, 6454–6463. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IκB kinase promotes tumorigenesis through inhibition of Forkhead FOXO3A. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Krishnan, R.K.; Nolte, H.; Sun, T.; Kaur, H.; Sreenivasan, K.; Looso, M.; Offermanns, S.; Kruger, M.; Swiercz, J.M. Quantitative analysis of the TNF-α-induced phosphoproteome reveals AEG-1/MTDH/LYRIC as an IKKβ substrate. Nat. Commun. 2015, 6, 6658. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xiang, J.; Lin, Y.; Ma, J.; Zhang, J.; Zhang, H.; Sun, J.; Danial, N.N.; Liu, J.; Lin, A. Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell 2013, 152, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NF-κ B activation in IKK-β-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in KRAS-induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41–55. [Google Scholar] [PubMed]
- Zaynagetdinov, R.; Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; McLoed, A.G.; Ausborn, J.A.; Polosukhin, V.V.; Connelly, L.; Zhou, W.; Fingleton, B.; et al. Epithelial nuclear factor-κB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene 2012, 31, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKβ- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of IKKb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKβ promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits PPARγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KRASG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-κ B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven Stat3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Wullaert, A.; Sasaki, Y.; Schmidt-Supprian, M.; Rajewsky, K.; Roskams, T.; Pasparakis, M. Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Investig. 2011, 121, 2781–2793. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Stratis, A.; Pasparakis, M.; Markur, D.; Knaup, R.; Pofahl, R.; Metzger, D.; Chambon, P.; Krieg, T.; Haase, I. Localized inflammatory skin disease following inducible ablation of IκB kinase 2 in murine epidermis. J. Investig. Dermatol. 2006, 126, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, K.S.; Walton, K.D.; Duncan, C.; Tjalkens, R.B. Spontaneous development of cutaneous squamous cell carcinoma in mice with cell-specific deletion of inhibitor of κB kinase 2. Comp. Med. 2017, 67, 407–415. [Google Scholar] [PubMed]
- Cornish, G.H.; Tung, S.L.; Marshall, D.; Ley, S.; Seddon, B.P. Tissue specific deletion of inhibitor of κB kinase 2 with OX40-CRE reveals the unanticipated expression from the OX40 locus in skin epidermis. PLoS ONE 2012, 7, e32193. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Bravo, A.; Suarez-Cabrera, C.; Alameda, J.P.; Casanova, M.L.; Lorz, C.; Segrelles, C.; Segovia, J.C.; Paramio, J.M.; Navarro, M.; et al. IKKβ-mediated resistance to skin cancer development is INK4A/ARF-dependent. Mol. Cancer Res. 2017, 15, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Cascallana, J.L.; Casanova, M.L.; Navarro, M.; Alameda, J.P.; Perez, P.; Bravo, A.; Ramirez, A. IKKβ overexpression leads to pathologic lesions in stratified epithelia and exocrine glands and to tumoral transformation of oral epithelia. Mol. Cancer Res. 2011, 9, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Tétreault, M.-P.; Weinblatt, D.; Ciolino, J.D.; Klein-Szanto, A.J.; Sackey, B.K.; Victor, C.T.-S.; Karakasheva, T.; Teal, V.; Katz, J.P. Esophageal expression of active IκB kinase-β in mice upregulates tumor necrosis factor and granulocyte macrophage colony-stimulating factor, promoting inflammation and angiogenesis. Gastroenterology 2016, 150, 1609–1619.e1611. [Google Scholar]
- Aleksic, T.; Baumann, B.; Wagner, M.; Adler, G.; Wirth, T.; Weber, C.K. Cellular immune reaction in the pancreas is induced by constitutively active IκB kinase-2. Gut 2007, 56, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Wagner, M.; Aleksic, T.; von Wichert, G.; Weber, C.K.; Adler, G.; Wirth, T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J. Clin. Investig. 2007, 117, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Shaked, H.; Hofseth, L.J.; Chumanevich, A.; Chumanevich, A.A.; Wang, J.; Wang, Y.; Taniguchi, K.; Guma, M.; Shenouda, S.; Clevers, H.; et al. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through INOS up-regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 14007–14012. [Google Scholar] [CrossRef] [PubMed]
- Leder, A.; Kuo, A.; Cardiff, R.D.; Sinn, E.; Leder, P. V-HA-RAS transgene abrogates the initiation step in mouse skin tumorigenesis: Effects of phorbol esters and retinoic acid. Proc. Natl. Acad. Sci. USA 1990, 87, 9178–9182. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Hsu, S.C.; Davidson, B.; Birrer, M.J.; Kohn, E.C.; Annunziata, C.M. Activation of NF-κB signaling by inhibitor of NF-κB kinase β increases aggressiveness of ovarian cancer. Cancer Res. 2010, 70, 4005–4014. [Google Scholar] [CrossRef] [PubMed]
- Kai, X.; Chellappa, V.; Donado, C.; Reyon, D.; Sekigami, Y.; Ataca, D.; Louissaint, A.; Mattoo, H.; Joung, J.K.; Pillai, S. IκB kinase β (IKBKB) mutations in lymphomas that constitutively activate canonical nuclear factor κB (NFκB) signaling. J. Biol. Chem. 2014, 289, 26960–26972. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- cBioPortal for Cancer Genomics. Available online: http://www.cbioportal.org (accessed on 18 October 2017).
- National Cancer Institute. Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov/ (accessed on 18 October 2017).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type of Tumor/IKKβ Modification | Phenotypic Effect | Proposed Mechanism | References |
|---|---|---|---|
| Lung Cancer | |||
| Lentiviral-mediated Ikbkb deletion in a lung model of adenocarcinoma expressing KrasG12D and shp53. | Attenuated tumor proliferation and significantly prolonged mouse survival. | Down-regulation of the NF-κB target TIMP-1 and ERK pathway; reduced cell proliferation. | [47] |
| Melanoma | |||
| Dox-induced Ikbkb deletion in melanocytes expressing HRasG12V in INK4A/ARF-null background. | Inhibition of melanoma tumor development. | p53-dependent cell cycle arrest and apoptosis. | [51] |
| Ikbkb deletion in myeloid cells in a mouse model injected with BRAFV600E /PTEN−/− melanoma cells. | Growth of cutaneous and lung melanoma tumors. | Myeloid IKKβ promotes antitumor immunity by modulating the chemokine CCL11 and the innate immune response. | [52] |
| Pancreatic Cancer | |||
| Cre-mediated Ikbkb deletion in a model expressing KrasG12D in the pancreas. | Reduced progression of pancreatic neoplasia. | Downregulation of inflammatory cytokines and chemokines; downregulation of Notch signaling; PPARG inhibition. | [53] |
| Cre-mediated Ikbkb deletion in a model expressing KrasG12D in the pancreas (also in INK4A/ARF null background). | Reduced formation of pancreatic neoplasia and of pancreatic ductal adenocarcinomas. | Inhibition of inflammation and NFκB activation. | [54] |
| Liver Cancer | |||
| Cre-mediated Ikbkb deletion in hepatocytes. | Enhanced DEN-induced hepatocarcinogenesis. | Increased cell death and compensatory proliferation mediated by increased ROS production and JNK1 activation. | [55,56] |
| Ikbkb deletion in initiated hepatocytes transplanted onto mice expressing PLAU in hepatocytes. | Enhanced formation of hepatocellular carcinomas. | Enhanced ROS production and STAT3 activation. | [57] |
| Cre-mediated Ikbkb deletion in hepatocytes and hematopoietic-derived Kupffer cells. | Decreased DEN-induced hepatocarcinogenesis and reduced hepatocyte regeneration. | Diminished induction of hepatic mitogens (IL-6, TNFα and HGF). | [55] |
| Intestinal Cancer | |||
| Cre-mediated Ikbkb deletion in intestinal epithelial cells. | Decreased tumor incidence in a colitis-associated cancer model. | Enhanced p53-independent apoptosis and defective Bcl-xL induction in tumor promotion. | [58] |
| Cre-mediated Ikbkb deletion in myeloid cells. | Decreased tumor incidence and size in a colitis-associated cancer model. | Reduced expression of proinflammatory mediators without effect on apoptosis. | [58] |
| Expression of constitutively active IKKβ in intestinal epithelial cells. | Spontaneous tumorigenesis and enhanced carcinogenesis induced by APC mutation or chemical treatments. | Activation of Wnt signaling and production of a pro-inflammatory intestinal microenvironment. | [59] |
| Ikbkb deletion in mesenchymal cells mediated by a constitutive ColVI-Cre transgene. | Protection against inflammation-induced intestinal carcinogenesis. | IKKβ in mecenchimal cell causes an increase in IL-6 production and STAT3 activation. | [60] |
| Ikbkb deletion in mesenchymal cells mediated by an inducible Col1a2Cre-ER transgene. | Stimulated intestinal proliferation, increased angiogenesis and promotion of colonic tumor growth. | IKKβ down-regulates TGFβ signaling and HGF secretion. | [61] |
| Non-Melanoma Skin Cancer | |||
| Constitutive K14-Cre mediated Ikbkb deletion in epidermal keratinocytes. | Severe inflammatory skin disease leading to death before postnatal day 10. | Unbalanced immune skin homeostasis, mediated by TNFα. | [62] |
| Inducible K14-CreER mediated Ikbkb deletion in epidermal keratinocytes. | Skin inflammation, hair follicle disruption and epidermal pseudoepitheliomatous hyperplasia but not tumor formation. | Ikbkb deletion leads to STAT3 and ERK1/2 activation. | [63] |
| Unexpected Ikbkb deletion in skin keratinocytes mediated by a GFAP-Cre transgene. | Skin hyperplasia, inflammation and development of SCCs in part of the mice. | Increased TNFα expression in lesions. | [64] |
| Unexpected Ikbkb deletion in skin keratinocytes mediated by a OX40-Cre transgene. | Hyperplasia and inflammatory skin lesions. | Increased TNFα expression in lesions and T lymphocyte activation. | [65] |
| IKKβ overexpression in epidermal keratinocytes by a K5-IKKβ transgene. | Resistance to tumor development in chemically-induced NMSC models. | Tumor-protective function of IKKβ is mediated by tumor suppressor proteins p16 and/or p19. | [66] |
| Oral and Esophageal Cancer | |||
| IKKβ overexpression in oral epithelial keratinocytes by a K5-IKKβ transgene. | Spontaneous oral tumoral lesions and increased malignancy after oral chemical carcinogenesis. | Enhanced oral inflammation with infiltration of granulocytes, macrophages and B lymphocytes. | [67] |
| Expression of constitutively active IKKβ in esophageal epithelia. | Esophagitis and increased angiogenesis in esophageal stroma. | Increased production and secretion of GM-CSF and TNF. | [68] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Page, A.; Navarro, M.; Suárez-Cabrera, C.; Bravo, A.; Ramirez, A. Context-Dependent Role of IKKβ in Cancer. Genes 2017, 8, 376. https://doi.org/10.3390/genes8120376
Page A, Navarro M, Suárez-Cabrera C, Bravo A, Ramirez A. Context-Dependent Role of IKKβ in Cancer. Genes. 2017; 8(12):376. https://doi.org/10.3390/genes8120376
Chicago/Turabian StylePage, Angustias, Manuel Navarro, Cristian Suárez-Cabrera, Ana Bravo, and Angel Ramirez. 2017. "Context-Dependent Role of IKKβ in Cancer" Genes 8, no. 12: 376. https://doi.org/10.3390/genes8120376
APA StylePage, A., Navarro, M., Suárez-Cabrera, C., Bravo, A., & Ramirez, A. (2017). Context-Dependent Role of IKKβ in Cancer. Genes, 8(12), 376. https://doi.org/10.3390/genes8120376