Dualistic Role of BARD1 in Cancer


Abstract
1. Introduction
2. Rare and Common Cancer-Associated Genetic Variants of BARD1
2.1. Rare Predisposing Variants of BARD1 in Cancer
2.2. Common Predisposing Variants of BARD1 in Cancer
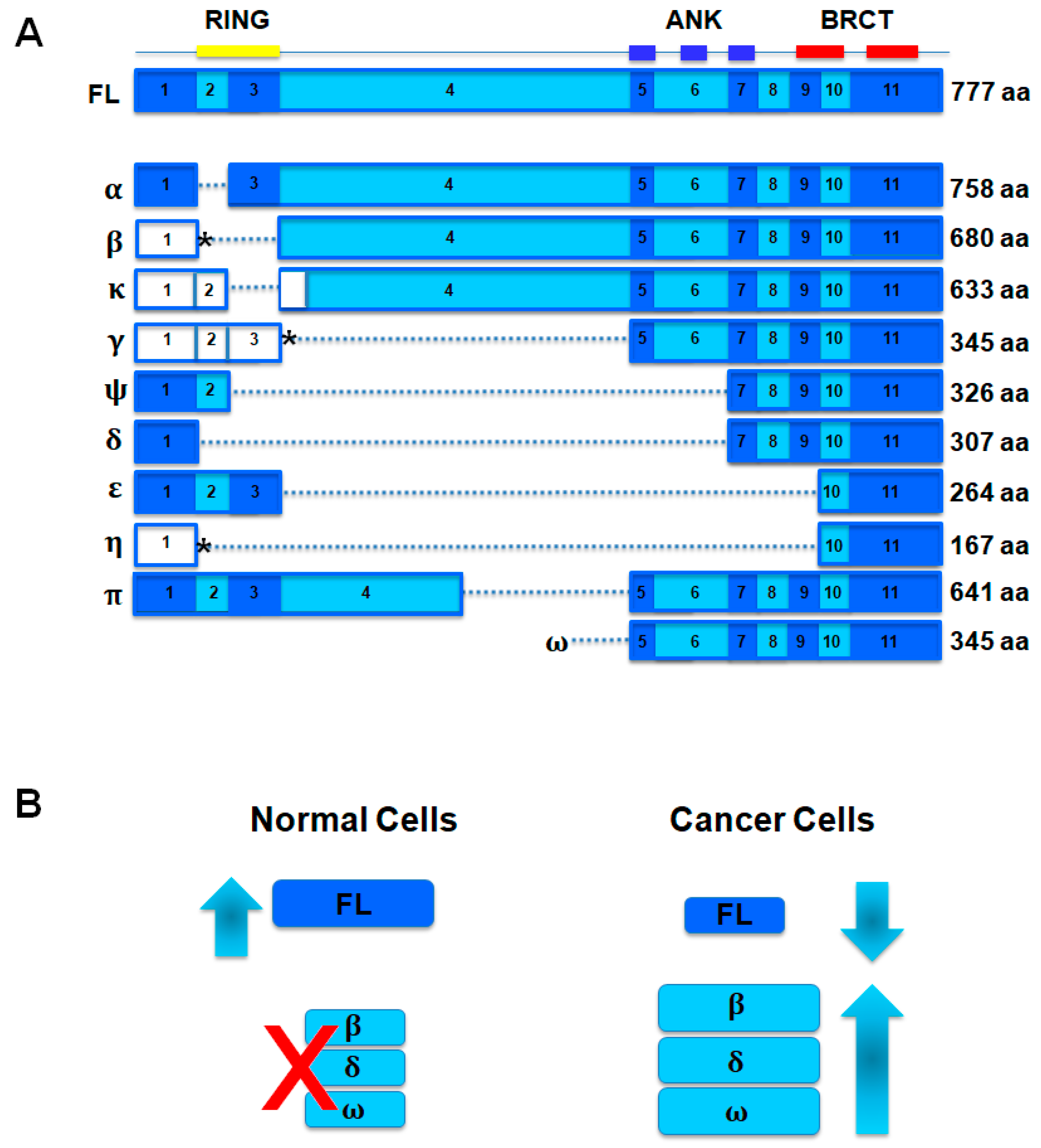
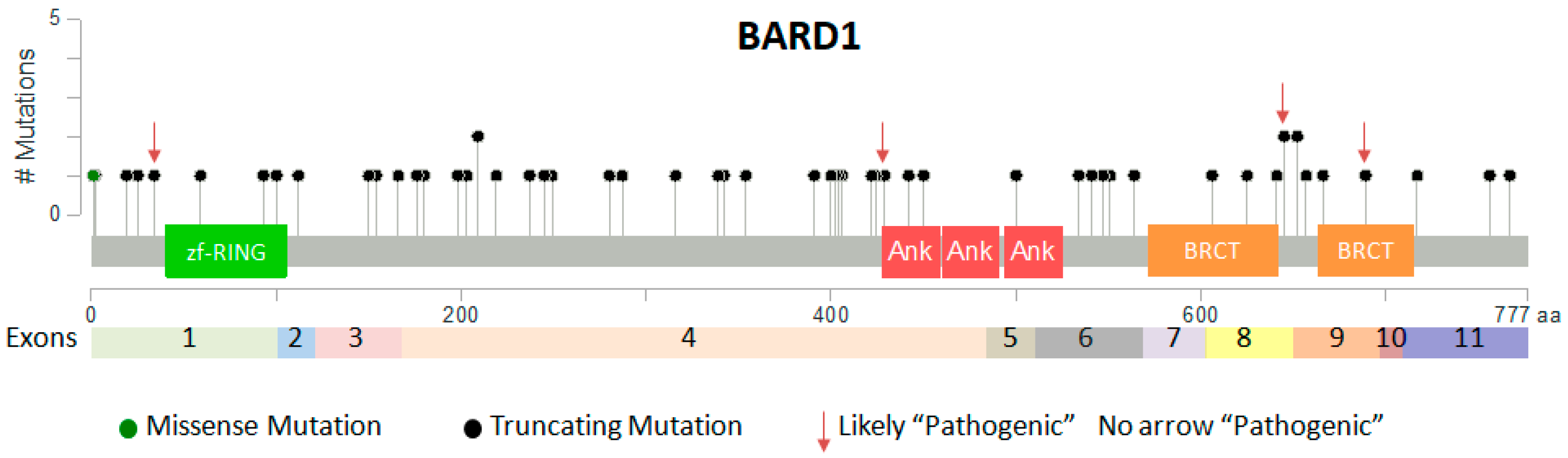
2.3. Somatic Mutations of BARD1 in Cancer
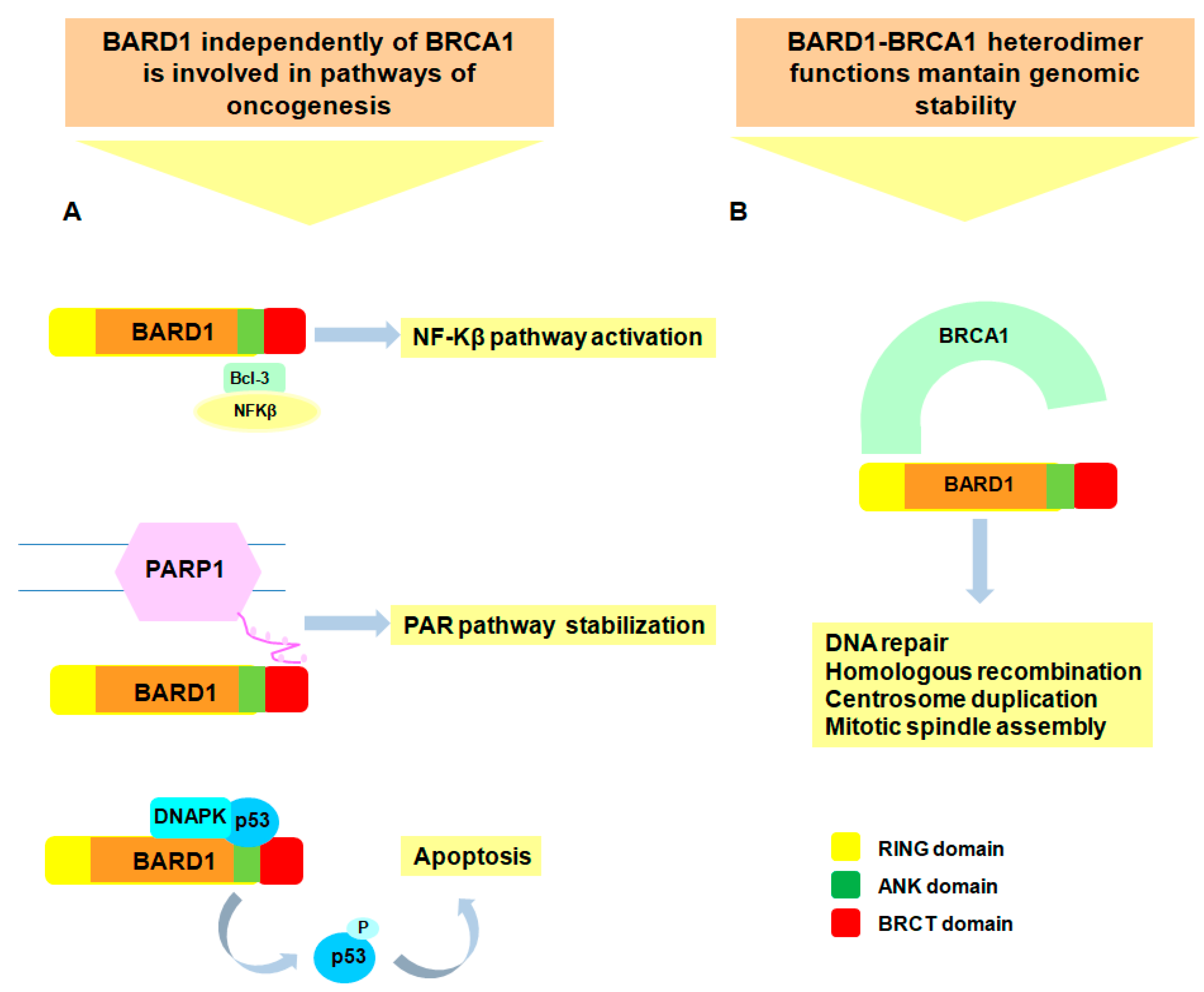
3. Biological Functions of BARD1 as Tumor Suppressor
4. Biological Functions of BARD1 as Oncogene
5. Summary and Future Perspectives
BARD1 as Possible Biomarker and Therapeutic Possibilities
Acknowledgments
Conflicts of Interest
References
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Meza, J.E.; King, M.C.; Klevit, R.E. BRCA1 RING domain cancer-predisposing mutations. Structural consequences and effects on protein-protein interactions. J. Biol. Chem. 2001, 276, 41399–41406. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Ratajska, M.; Pilyugin, M. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int. J. Biochem. Cell Biol. 2016, 72, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Jefford, C.E. Is there more to BARD1 than BRCA1? Nat. Rev. Cancer 2006, 6, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Soriano, J.V.; Vaudan, G.; Montesano, R.; Sappino, A.P. In Vitro repression of Brca1-associated RING domain gene, Bard1, induces phenotypic changes in mammary epithelial cells. J. Cell Biol. 1998, 143, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Ayi, T.C.; Tsan, J.T.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Conservation of function and primary structure in the BRCA1-associated RING domain (BARD1) protein. Oncogene 1998, 17, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Chen, J.; Fox, E.A.; Green, J.B.; Livingston, D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA 2001, 98, 12078–12083. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J.; Martin, J.S.; Polanowska, J.; Hill, D.E.; Gartner, A.; Vidal, M. BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr. Biol. 2004, 14, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lafarge, S.; Montane, M.H. Characterization of Arabidopsis thaliana ortholog of the human breast cancer susceptibility gene 1: AtBRCA1, strongly induced by gamma rays. Nucleic Acids Res. 2003, 31, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Shakya, R.; Szabolcs, M.; McCarthy, E.; Ospina, E.; Basso, K.; Nandula, S.; Murty, V.; Baer, R.; Ludwig, T. The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. USA 2008, 105, 7040–7045. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.E.; Celebi, J.T.; Baer, R.; Ludwig, T. Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol. Cell. Biol. 2003, 23, 5056–5063. [Google Scholar] [CrossRef] [PubMed]
- Ghimenti, C.; Sensi, E.; Presciuttini, S.; Brunetti, I.M.; Conte, P.; Bevilacqua, G.; Caligo, M.A. Germline mutations of the BRCA1-associated RING domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer 2002, 33, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ishitobi, M.; Miyoshi, Y.; Hasegawa, S.; Egawa, C.; Tamaki, Y.; Monden, M.; Noguchi, S. Mutational analysis of BARD1 in familial breast cancer patients in Japan. Cancer Lett. 2003, 200, 1–7. [Google Scholar] [CrossRef]
- Karppinen, S.M.; Heikkinen, K.; Rapakko, K.; Winqvist, R. Mutation screening of the BARD1 gene: Evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J. Med. Genet. 2004, 41, e114. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Vlastos, A.T.; Pelte, M.F.; Caligo, M.A.; Bianco, A.; Krause, K.H.; Laurent, G.J.; Irminger-Finger, I. Aberrant expression of BARD1 in breast and ovarian cancers with poor prognosis. Int. J. Cancer 2006, 118, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I. BARD1, a possible biomarker for breast and ovarian cancer. Gynecol. Oncol. 2010, 117, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar] [CrossRef]
- Sporn, J.C.; Hothorn, T.; Jung, B. BARD1 expression predicts outcome in colon cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5451–5462. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Ryser, S.; Dizin, E.; Jefford, C.E.; Delaval, B.; Gagos, S.; Christodoulidou, A.; Krause, K.H.; Birnbaum, D.; Irminger-Finger, I. Distinct roles of BARD1 isoforms in mitosis: Full-Length BARD1 mediates Aurora B degradation, cancer-associated BARD1β scaffolds Aurora B and BRCA2. Cancer Res. 2009, 69, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Irminger-Finger, I. Long non-coding RNA and microRNAs might act in regulating the expression of BARD1 mRNAs. Int. J. Biochem. Cell Biol. 2014, 54, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.H.; Du, F.; Tsan, J.T.; Jin, Y.; Phung, A.; Spillman, M.A.; Massa, H.F.; Muller, C.Y.; Ashfaq, R.; Mathis, J.M.; et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998, 7, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Stacey, S.N.; Sulem, P.; Johannsson, O.T.; Helgason, A.; Gudmundsson, J.; Kostic, J.P.; Kristjansson, K.; Jonsdottir, T.; Sigurdsson, H.; Hrafnkelsson, J.; et al. The BARD1 Cys557Ser variant and breast cancer risk in Iceland. PLoS Med. 2006, 3, e217. [Google Scholar] [CrossRef] [PubMed]
- Guenard, F.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Durocher, F.; BRCAs, I. Genetic sequence variations of BRCA1-interacting genes AURKA, BAP1, BARD1 and DHX9 in French Canadian families with high risk of breast cancer. J. Hum. Genet. 2009, 54, 152–161. [Google Scholar] [CrossRef] [PubMed]
- De Brakeleer, S.; De Greve, J.; Loris, R.; Janin, N.; Lissens, W.; Sermijn, E.; Teugels, E. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 2010, 31, E1175–E1185. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Ratajska, M.; Antoszewska, E.; Piskorz, A.; Brozek, I.; Borg, A.; Kusmierek, H.; Biernat, W.; Limon, J. Cancer predisposing BARD1 mutations in breast-ovarian cancer families. Breast Cancer Res. Treat. 2012, 131, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ratajska, M.; Matusiak, M.; Kuzniacka, A.; Wasag, B.; Brozek, I.; Biernat, W.; Koczkowska, M.; Debniak, J.; Sniadecki, M.; Kozlowski, P.; et al. Cancer predisposing BARD1 mutations affect exon skipping and are associated with overexpression of specific BARD1 isoforms. Oncol. Rep. 2015, 34, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, V.A.; Formicola, D.; Pignataro, P.; Cimmino, F.; Calabrese, F.M.; Mora, J.; Esposito, M.R.; Pantile, M.; Zanon, C.; De Mariano, M.; et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 2016, 7, 21840–21852. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Silversides, C.K.; Lionel, A.C.; Costain, G.; Merico, D.; Migita, O.; Liu, B.; Yuen, T.; Rickaby, J.; Thiruvahindrapuram, B.; Marshall, C.R.; et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012, 8, e1002843. [Google Scholar] [CrossRef] [PubMed]
- Faingold, R.; Babyn, P.S.; Yoo, S.J.; Dipchand, A.I.; Weitzman, S. Neuroblastoma with atypical metastases to cardiac and skeletal muscles: MRI features. Pediatr. Radiol. 2003, 33, 584–586. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Lipshultz, S.E.; Lipsitz, S.R.; Colan, S.D.; Diller, L. Association between congenital cardiovascular malformations and neuroblastoma. J. Pediatr. 2004, 144, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Caminsky, N.G.; Mucaki, E.J.; Perri, A.M.; Lu, R.; Knoll, J.H.; Rogan, P.K. Prioritizing variants in complete hereditary breast and ovarian cancer genes in patients lacking known BRCA mutations. Hum. Mutat. 2016, 37, 640–652. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.D.; Conkrite, K.L.; Chang, X.; Capasso, M.; Vaksman, Z.; Oldridge, D.A.; Zachariou, A.; Horn, M.; Diamond, M.; Hou, C.; et al. Common variants upstream of MLF1 at 3q25 and within CPZ at 4p16 associated with neuroblastoma. PLoS Genet. 2017, 13, e1006787. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; McDaniel, L.D.; Cimmino, F.; Cirino, A.; Formicola, D.; Russell, M.R.; Raman, P.; Cole, K.A.; Diskin, S.J. The functional variant rs34330 of CDKN1B is associated with risk of neuroblastoma. J. Cell. Mol. Med. 2017, 21, 3224–3230. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.; Cimmino, F.; Acierno, G.; Totaro, F.; Petrosino, G.; Pezone, L.; Diamond, M.; McDaniel, L.; Hakonarson, H.; et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. 2014, 74, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Diamond, M.; Oldridge, D.A.; Conkrite, K.; Bosse, K.R.; Russell, M.R.; Iolascon, A.; Hakonarson, H.; Devoto, M.; et al. Rare variants in TP53 and susceptibility to neuroblastoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Diskin, S.J.; Zhang, H.; Attiyeh, E.F.; Winter, C.; Hou, C.; Schnepp, R.W.; Diamond, M.; Bosse, K.; Mayes, P.A.; et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 2011, 469, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Mosse, Y.P.; Bradfield, J.P.; Hou, C.; Monni, S.; Scott, R.H.; Asgharzadeh, S.; Attiyeh, E.F.; Diskin, S.J.; Laudenslager, M.; et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 2008, 358, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Latorre, V.; Diskin, S.J.; Diamond, M.A.; Zhang, H.; Hakonarson, H.; Maris, J.M.; Devoto, M. Replication of neuroblastoma SNP association at the BARD1 locus in African-Americans. Cancer Epidemiol. Biomark. Prev. 2012, 21, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zou, Y.; Zhu, J.; Zeng, X.; Yang, T.; Wang, F.; He, J.; Xia, H. The Association between GWAS-identified BARD1 Gene SNPs and Neuroblastoma Susceptibility in a Southern Chinese Population. Int. J. Med. Sci. 2016, 13, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F. BARD1 locus of neuroblastoma susceptibility; Università di Napoli Federico II: Naples, Italy, 2017. [Google Scholar]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen, L.B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Calabrese, F.M.; Iolascon, A.; Mellerup, E. Combinations of genetic data in a study of neuroblastoma risk genotypes. Cancer Genet. 2014, 207, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhu, J.; Xiong, S.W.; Jia, W.; Zhao, Z.; Zhu, S.B.; Hu, J.H.; Wang, F.H.; Xia, H.; He, J.; et al. BARD1 gene polymorphisms confer nephroblastoma susceptibility. EBioMedicine 2017, 16, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hormazabal, P.; Reyes, J.M.; Blanco, R.; Bravo, T.; Carrera, I.; Peralta, O.; Gomez, F.; Waugh, E.; Margarit, S.; Ibanez, G.; et al. The BARD1 Cys557Ser variant and risk of familial breast cancer in a South-American population. Mol. Biol. Rep. 2012, 39, 8091–8098. [Google Scholar] [CrossRef] [PubMed]
- Jakubowska, A.; Cybulski, C.; Szymanska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Debniak, T.; Gorski, B.; Kowalska, E.; Narod, S.A.; et al. BARD1 and breast cancer in Poland. Breast Cancer Res. Treat. 2008, 107, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Marquart, L.; McGuffog, L.; Healey, S.; Sinilnikova, O.; Wan, F.; Chen, X.; Beesley, J.; Singer, C.F.; Dressler, A.C.; et al. Common genetic variation at BARD1 is not associated with breast cancer risk in BRCA1 or BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.P.; Zhang, Y.; Ma, W.L.; He, X.F.; Wang, W.; Yu, H.L.; Guo, Y.B.; Zheng, W.L. Lack of association between BARD1 Cys557Ser variant and breast cancer risk: A meta-analysis of 11,870 cases and 7687 controls. J. Cancer Res. Clin. Oncol. 2011, 137, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Johnatty, S.E.; Beesley, J.; Chen, X.; Hopper, J.L.; Southey, M.C.; Giles, G.G.; Goldgar, D.E.; Chenevix-Trench, G.; Spurdle, A.B. The BARD1 Cys557Ser polymorphism and breast cancer risk: An Australian case-control and family analysis. Breast Cancer Res. Treat. 2009, 115, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. 3), S3. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Machida, Y.; Sankaran, S.; Elias, J.E.; Griffin, K.; Schlegel, B.P.; Gygi, S.P.; Parvin, J.D. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol. Cell. Biol. 2004, 24, 8457–8466. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.C.; Doan, T.P.; White, R.L. Identification of a gamma-tubulin-binding domain in BRCA1. Cancer Res. 2001, 61, 7713–7718. [Google Scholar] [PubMed]
- Westermark, U.K.; Reyngold, M.; Olshen, A.B.; Baer, R.; Jasin, M.; Moynahan, M.E. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol. 2003, 23, 7926–7936. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Groen, A.C.; Prokhorova, T.; Gerson, R.; White, E.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006, 127, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Manley, J.L. Deregulation of poly(A) polymerase interferes with cell growth. Mol. Cell. Biol. 1998, 18, 5010–5020. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, F.E.; Manley, J.L. The BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell 2001, 104, 743–753. [Google Scholar] [CrossRef]
- Dechend, R.; Hirano, F.; Lehmann, K.; Heissmeyer, V.; Ansieau, S.; Wulczyn, F.G.; Scheidereit, C.; Leutz, A. The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene 1999, 18, 3316–3323. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Jefford, C.E.; Berardi, P.; Wu, J.Y.; Cartier, L.; Krause, K.H.; Irminger-Finger, I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 2005, 24, 3726–3736. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Leung, W.C.; Li, J.; Dubois-Dauphin, M.; Harb, J.; Feki, A.; Jefford, C.E.; Soriano, J.V.; Jaconi, M.; Montesano, R.; et al. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol. Cell 2001, 8, 1255–1266. [Google Scholar] [CrossRef]
- Jefford, C.E.; Feki, A.; Harb, J.; Krause, K.H.; Irminger-Finger, I. Nuclear-cytoplasmic translocation of BARD1 is linked to its apoptotic activity. Oncogene 2004, 23, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D.; Xu, H.; Baer, R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 2004, 279, 33909–33918. [Google Scholar] [CrossRef] [PubMed]
- Hayami, R.; Sato, K.; Wu, W.; Nishikawa, T.; Hiroi, J.; Ohtani-Kaneko, R.; Fukuda, M.; Ohta, T. Down-regulation of BRCA1-BARD1 ubiquitin ligase by CDK2. Cancer Res. 2005, 65, 6–10. [Google Scholar] [PubMed]
- Smith, P.D.; Crossland, S.; Parker, G.; Osin, P.; Brooks, L.; Waller, J.; Philp, E.; Crompton, M.R.; Gusterson, B.A.; Allday, M.J.; et al. Novel p53 mutants selected in BRCA-associated tumours which dissociate transformation suppression from other wild-type p53 functions. Oncogene 1999, 18, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.A.; Nichol, K.; Ozcelik, H.; Knight, J.; Done, S.J.; Goodwin, P.J.; Andrulis, I.L. Frequency of p53 mutations in breast carcinomas from Ashkenazi Jewish carriers of BRCA1 mutations. J. Natl. Cancer Inst. 1999, 91, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ryser, S.; Dizin, E.; Pils, D.; Krainer, M.; Jefford, C.E.; Bertoni, F.; Zeillinger, R.; Irminger-Finger, I. Oncogenic BARD1 isoforms expressed in gynecological cancers. Cancer Res. 2007, 67, 11876–11885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Bianco, A.; Malkinson, A.M.; Leoni, V.P.; Frau, G.; De Rosa, N.; Andre, P.A.; Versace, R.; Boulvain, M.; Laurent, G.J.; et al. BARD1: An independent predictor of survival in non-small cell lung cancer. Int. J. Cancer 2012, 131, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Jefford, C.E.; Durand, P.; Harb, J.; Lucas, H.; Krause, K.H.; Irminger-Finger, I. BARD1 expression during spermatogenesis is associated with apoptosis and hormonally regulated. Biol. Reprod. 2004, 71, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, M.; Wu, W.; Nishikawa, H.; Hayami, R.; Oyake, D.; Yabuki, Y.; Fukuda, M.; Ohta, T. A truncated splice variant of human BARD1 that lacks the RING finger and ankyrin repeats. Cancer Lett. 2006, 233, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cohen, M.; Wu, J.; Sow, M.H.; Nikolic, B.; Bischof, P.; Irminger-Finger, I. Identification of BARD1 splice-isoforms involved in human trophoblast invasion. Int. J. Biochem. Cell Biol. 2007, 39, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Tembe, V.; Henderson, B.R. BARD1 translocation to mitochondria correlates with Bax oligomerization, loss of mitochondrial membrane potential and apoptosis. J. Biol. Chem. 2007, 282, 20513–20522. [Google Scholar] [CrossRef] [PubMed]
- Dizin, E.; Irminger-Finger, I. Negative feedback loop of BRCA1-BARD1 ubiquitin ligase on estrogen receptor α stability and activity antagonized by cancer-associated isoform of BARD1. Int. J. Biochem. Cell Biol. 2010, 42, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Andre, P.A.; Ratajska, M.; Kuzniacka, A.; Limon, J.; Tournier, B.B.; Colas, J.; Laurent, G.; Irminger-Finger, I. Antagonizing functions of BARD1 and its alternatively spliced variant BARD1δ in telomere stability. Oncotarget 2017, 8, 9339–9353. [Google Scholar] [CrossRef] [PubMed]
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC inhibitors repress BARD1 isoform expression in acute myeloid leukemia cells via activation of miR-19a and/or b. PLoS ONE 2013, 8, e83018. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J. Genetics and genomics of neuroblastoma. Cancer Treat. Res. 2010, 155, 65–84. [Google Scholar] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Ozden, O.; Bishehsari, F.; Bauer, J.; Park, S.H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.J.; Yazici, C.; Krett, N.; et al. Expression of an oncogenic BARD1 splice variant impairs homologous recombination and predicts response to PARP-1 inhibitor therapy in colon cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Descloux, P.; Andre, P.A.; Laszlo, V.; Dome, B.; Hegedus, B.; Sardy, S.; Janes, S.; Bianco, A.; Laurent, G.J.; et al. BARD1 serum autoantibodies for the detection of lung cancer. PLoS ONE 2017, 12, e0182356. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variation ID | GRCh37 Location | Ref. | Alt | Mutation Type | Protein Change | dbSNP | Frequency in ExAC Database | Condition(s) | Clinical Significance (Last Reviewed) |
|---|---|---|---|---|---|---|---|---|---|
| 237823 | . | . | . | deletion | . | . | . | Familial cancer of breast | Pathogenic (Last reviewed: 10 December 2015) |
| 230523 | 215593433–215593434 | TG | - | frameshift deletion | V767fs | rs750413473 | 0.00006 | Familial cancer of breast|not specified|Hereditary cancer-predisposing syndrome | Conflicting interpretations of pathogenicity (Last reviewed: 18 August 2016) |
| 185366 | 215593466 | G | A | nonsense | W756 * | rs786202118 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 20 June 2014) |
| 187542 | 215593585–215593586 | CA | - | frameshift deletion | I717fs | rs786203811 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 10 December 2014) |
| 422826 | 215593671 | A | AA | frameshift duplication | D689fs | . | . | not provided | Likely pathogenic (Last reviewed: 29 November 2016) |
| 265365 | 215593734 | A | C | splice acceptor | . | rs876658260 | . | not provided | Likely pathogenic (Last reviewed: 28 June 2016) |
| 229902 | 215593734 | A | T | splice acceptor | . | rs876658260 | . | Hereditary cancer-predisposing syndrome | Likely pathogenic (Last reviewed: 17 November 2015) |
| 182051 | 215595140 | C | T | nonsense | Q666 * | rs730881422 | . | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic/Likely pathogenic (Last reviewed: 13 January 2017) |
| 187445 | 215595166 | C | - | frameshift deletion | P657fs | rs786203739 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 3 March 2016) |
| 430952 | 215595182–215595201 | frameshift duplication | Q652fs | . | . | Familial cancer of breast | Pathogenic (Last reviewed: 13 April 2017) | ||
| 265510 | 215595182–215595201 | frameshift deletion | C645fs | rs886039589 | . | not provided | Likely pathogenic (Last reviewed: 3 December 2015) | ||
| 127725 | 215595182–215595201 | frameshift duplication | Q652fs | rs587780024 | . | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic/Likely pathogenic (Last reviewed: 13 April 2017) | ||
| 142499 | 215595203–215595204 | AT | - | frameshift deletion | C645fs | rs587782504 | . | Hereditary cancer-predisposing syndrome | Pathogenic/Likely pathogenic (Last reviewed: 20 November 2015) |
| 141702 | 215595215 | C | T | nonsense | R641 * | rs587781948 | 0.00001 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 4 October 2016) |
| 232108 | 215595234 | A | - | splice acceptor | . | rs876659560 | . | Hereditary cancer-predisposing syndrome | Likely pathogenic (Last reviewed: 29 May 2015) |
| 219763 | 215595234 | A | T | splice acceptor | . | rs864622239 | . | Familial cancer of breast | Likely pathogenic (Last reviewed: 8 August 2015) |
| 233167 | 215609790 | G | T | splice donor | . | rs876660237 | . | Hereditary cancer-predisposing syndrome | Likely pathogenic (Last reviewed: 6 August 2015) |
| 232127 | 215609822 | T | - | frameshift deletion | L625fs | rs876659572 | . | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 26 July 2016) |
| 143017 | 215609876–215609877 | AT | - | frameshift deletion | H606fs | rs587782897 | . | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 8 June 2016) |
| 245801 | 215609884 | G | A | splice acceptor | . | rs879253952 | . | not provided | Pathogenic (Last reviewed: 9 June 2015) |
| 246449 | 215609885–215609893 | - | splice acceptor | . | rs879254264 | . | not provided | Likely pathogenic (Last reviewed: 7 March 2016) | |
| 232643 | 215610445 | G | A | splice donor | . | rs876659894 | . | Hereditary cancer-predisposing syndrome | Likely pathogenic (Last reviewed: 30 June 2015) |
| 127720 | 215610566 | C | T | nonsense | Q564* | rs587780021 | 0.00005 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 3 January 2017) |
| 406791 | 215617170 | G | C | splice donor | . | rs1060501310 | . | Familial cancer of breast | Likely pathogenic (Last reviewed: 2 August 2016) |
| 141384 | 215617196 | C | G | nonsense | S551 * | rs587781707 | . | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 29 December 2016) |
| 230172 | 215617209 | G | T | nonsense | E547 * | rs876658429 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 28 January 2015) |
| 406768 | 215617214–215617248 | frameshift indel | T534fs | rs1064792931 | . | Familial cancer of breast | Pathogenic (Last reviewed: 22 September 2016) | ||
| 379750 | 215617226 | C | A | nonsense | S541 * | rs777937955 | . | not provided | Pathogenic (Last reviewed: 14 May 2015) |
| 406776 | 215632275 | - | A | frameshift duplication | D500fs | . | . | Familial cancer of breast | Pathogenic (Last reviewed: 13 August 2016) |
| 421820 | 215633955 | - | G | splice donor duplication | . | . | . | not provided | Likely pathogenic (Last reviewed: 4 August 2016) |
| 233414 | 215634002 | - | A | frameshift duplication | N450fs | rs876660390 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 17 August 2015) |
| 406748 | 215634026 | C | - | frameshift deletion | P442fs | rs1060501287 | . | Familial cancer of breast|not provided | Pathogenic/Likely pathogenic (Last reviewed: 20 September 2016) |
| 243116 | 215645314 | A | - | frameshift deletion | E429fs | rs879253879 | . | not provided | Likely pathogenic (Last reviewed: 11 December 2015) |
| 234190 | 215645328 | A | - | frameshift deletion | R424fs | rs876660911 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 2 October 2015) |
| 187182 | 215645331–215645334 | GTG | ATG | frameshift indel | V422fs | rs786203533 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 10 November 2014) |
| 229677 | 215645382 | C | T | nonsense | R406 * | rs377153250 | 0.00003 | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 11 November 2014) |
| 142734 | 215645386 | C | G | nonsense | Y404 * | rs587782681 | . | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 13 October 2016) |
| 379884 | 215645393 | C | G | nonsense | S402 * | rs796666047 | . | not provided | Pathogenic (Last reviewed: 1 June 2015) |
| 187646 | 215645400 | A | - | frameshift deletion | S400fs | rs786203891 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 17 December 2014) |
| 237814 | 215645426 | C | G | nonsense | S391 * | rs878853995 | . | Familial cancer of breast | Pathogenic (Last reviewed: 24 February 2016) |
| 185916 | 215645537 | C | G | nonsense | S354 * | rs786202559 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 24 March 2016) |
| 232902 | 215645575 | G | - | frameshift deletion | S342fs | rs876660061 | . | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 21 July 2015) |
| 419156 | 215645584 | C | - | frameshift deletion | S339fs | rs1064793682 | . | not provided | Pathogenic (Last reviewed: 1 June 2015) |
| 141412 | 215645651 | T | G | nonsense | L316 * | rs587781728 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 7 April 2014) |
| 185015 | 215645737–215645738 | AG | - | frameshift deletion | E287fs | rs786201868 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 29 May 2014) |
| 232418 | 215645759–215645760 | TT | - | frameshift deletion | L280fs | rs876659752 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 15 June 2015) |
| 406757 | 215645853 | - | A | frameshift duplication | I249fs | . | . | Familial cancer of breast | Pathogenic (Last reviewed: 24 October 2016) |
| 141005 | 215645865 | C | T | nonsense | Q245 * | rs587781430 | 0.00001 | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 8 March 2017) |
| 230107 | 215645889 | C | T | nonsense | Q237 * | rs587780035 | . | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 11 July 2016) |
| 141342 | 215645938–215645941 | TTTA | - | frameshift deletion | R219fs | rs587781671 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 23 February 2014) |
| 219726 | 215645970–215645971 | AA | - | frameshift deletion | K209fs | rs864622223 | . | Familial cancer of breast|not provided | Pathogenic/Likely pathogenic (Last reviewed: 4 December 2015) |
| 127742 | 215645975 | - | A | frameshift deletion | K209fs | rs587780033 | . | Familial cancer of breast|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 18 November 2016) |
| 182042 | 215645991 | G | T | nonsense | G203 * | rs730881415 | . | not provided | Pathogenic (Last reviewed: 25 September 2014) |
| 233965 | 215646006 | G | - | frameshift deletion | A198fs | rs876660761 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 15 September 2015) |
| 186576 | 215646058–215646059 | AT | - | frameshift deletion | Y180fs | rs779427628 | 0.00001 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 13 December 2016) |
| 406761 | 215646072 | C | T | nonsense | Q176 * | rs776851287 | . | Familial cancer of breast | Pathogenic (Last reviewed: 26 May 2016) |
| 185846 | 215646102 | C | T | nonsense | Q166 * | rs786202500 | . | not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 4 April 2016) |
| 230344 | 215646138–215646141 | - | AAAG | frameshift duplication | V154fs | rs772486760 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 2 February 2015) |
| 182036 | 215646150 | C | T | nonsense | R150 * | rs730881411 | 0.00001 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 29 July 2016) |
| 185139 | 215657051 | C | T | nonsense | R112 * | rs758972589 | 0.00001 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 27 May 2016) |
| 185079 | 215657087 | C | T | nonsense | Q100 * | rs786201912 | . | Familial cancer of breast|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 13 December 2016) |
| 230447 | 215657108 | C | T | nonsense | Q93 * | rs876658571 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 23 February 2015) |
| 231019 | 215657170 | G | A | splice acceptor | . | rs876658905 | . | Hereditary cancer-predisposing syndrome | Likely pathogenic (Last reviewed: 26 March 2015) |
| 371931 | 215661823–215661824 | AG | - | frameshift deletion | E59fs | rs1057517589 | . | Familial cancer of breast | Pathogenic/Likely pathogenic (Last reviewed: 16 November 2016) |
| 246176 | 215661842 | G | T | splice acceptor | . | rs879254139 | . | not provided | Likely pathogenic (Last reviewed: 3 December 2015) |
| 246476 | 215674192 | G | A | nonsense | W34 * | rs879254280 | . | not provided | Likely pathogenic (Last reviewed: 8 March 2016) |
| 231232 | 215674224–215674225 | frameshift indel | A25fs | rs876659040 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 25 March 2015) | ||
| 233594 | 215674239 | G | T | nonsense | E19 * | rs752514155 | 0.00002 | Familial cancer of breast|not provided|Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 28 August 2016) |
| 232926 | 215674271–215674290 | frameshift deletion | P2fs | rs876660077 | . | Hereditary cancer-predisposing syndrome | Pathogenic (Last reviewed: 10 July 2015) | ||
| 127739 | 215674291 | G | A | missense | M1I | rs587780031 | . | not provided | Pathogenic (Last reviewed: 5 November 2013) |
| 154206 | 190300875–242783384 | . | . | copy number gain (2q32.2-37.3) | . | nssv3395386 | . | Hypospadias | Pathogenic (Last reviewed: 18 March 2014) |
| 152738 | 193803552–216569775 | . | . | copy number loss (2q32.3-35) | . | nssv1608180 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 14 January 2013) |
| 150620 | 181378520–225167565 | . | . | copy number gain (2q31.3-36.1) | . | nssv1604018 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 25 July 2011) |
| 146683 | 211444400–243059659 | . | . | copy number gain (2q34-37.3) | . | nssv706486 | . | Coarctation of aorta | Pathogenic (Last reviewed: 25 February 2011) |
| 59164 | 213479146–227985946 | . | . | copy number gain (2q34-36) | . | nssv578841 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 12 August 2011) |
| 59160 | 193987039–242014395 | . | . | copy number gain (2q32.3-37.3) | . | nssv578835 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 12 August 2011) |
| 59159 | 191175462–242834921 | . | . | copy number gain (2q32.2-37.3) | . | nssv578834 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 12 August 2011) |
| 59158 | 189682921–243007359 | . | . | copy number gain (2q32.2-37.3) | . | nssv578833 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 12 August 2011) |
| 57419 | 195763507–237382556 | . | . | copy number gain (2q32.3-37.3) | . | nssv578837 | . | Developmental delay AND/OR other significant developmental or morphological phenotypes | Pathogenic (Last reviewed: 12 August 2011) |
| Position | Sequence Ontology | Protein Sequence Change | CHASM p-Value | VEST p-Value | ID dbSNP | Frequency in ExAC Database | COSMIC Variant Count in Tissues (n) |
|---|---|---|---|---|---|---|---|
| 214769310 | SG | R123 * | 0.1290 | rs369986649 | 0.00 | large_intestine (1); lung (1); endometrium (1); skin(2) | |
| 214781072 | MS | E268K | 0.2724 | 0.4382 | 0.00 | cervix(1); liver(2) | |
| 214728840 | MS | A724T | 0.0462 | 0.0145 | 0.00 | large_intestine(2) | |
| 214730417 | MS | E665D | 0.3204 | 0.5076 | 0.00 | liver(2) | |
| 214728936 | MS | I692F | 0.4718 | 0.2635 | 0.00 | liver(2) | |
| 214781454 | MS | K140N | 0.5620 | 0.1730 | rs758749603 | 0.000008 | large_intestine(1); endometrium(1) |
| 214781285 | SG | K197 * | 0.2026 | 0.00 | liver(2) | ||
| 214781251 | FD | K208RKL * | 0.0588 | 0.000017 | large_intestine(1) | ||
| 214780799 | SY | L359L | 0.00 | esophagus(2) | |||
| 214730452 | MS | P654S | 0.0970 | 0.6800 | 0.00 | skin(2) | |
| 214792396 | MS | P89A | 0.2542 | 0.0534 | 0.00 | pancreas(2) | |
| 214728907 | MS | Q701H | 0.1730 | 0.1709 | 0.00 | esophagus(1) | |
| 214781390 | MS | S162A | 0.5712 | 0.2689 | 0.00 | hematopoietic_and_lymphoid_tissue(2) | |
| 214780783 | MS | S364L | 0.4934 | 0.7981 | rs200168806 | 0.00 | esophagus(2) |
| 214780784 | MS | S364T | 0.7164 | 0.3940 | rs201292946 | 0.00 | esophagus(2) |
| 214780955 | MS | Y307N | 0.6760 | 0.5594 | 0.00 | esophagus(1) | |
| 214769314 | SS | 0.0374 | 0.00 | ||||
| 214781510 | SS | 0.1037 | 0.00 | ||||
| 214728990 | SG | G674 * | 0.0577 | 0.00 | |||
| 214745084 | SG | W629 * | 0.0191 | rs747446711 | 0.000008 | lung(1) | |
| 214780882 | MS | P331R | 0.6070 | 0.2104 | 0.00 | autonomic_ganglia(1) | |
| 214780655 | MS | V407M | 0.2596 | 0.8458 | 0.00 | biliary_tract(1) | |
| 214781361 | ^ FD | see footnote | 0.2006 | 0.000016 | biliary_tract(1) | ||
| 214781136 | SY | P246P | rs587780859 | 0.000096 | bone(1) | ||
| 214781165 | MS | Q237E | 0.4816 | 0.8826 | rs587780035 | 0.000096 | bone(1) |
| 214745777 | SY | L585L | 0.00 | breast(1) | |||
| 214745791 | MS | Q581K | 0.3898 | 0.0927 | 0.00 | breast(1) | |
| 214792394 | SY | P89P | rs756165637 | 0.000008 | breast(1) | ||
| 214781066 | MS | E270K | 0.3180 | 0.3405 | 0.00 | cervix(1) | |
| 214781210 | MS | A222S | 0.8046 | 0.7358 | 0.00 | cervix(1) | |
| 214781384 | SG | Q164 * | 0.1407 | 0.00 | endometrium(1) | ||
| 214781306 | MS | D190Y | 0.6800 | 0.5193 | rs369561166 | 0.000008 | endometrium(1) |
| 214781158 | MS | L239R | 0.4934 | 0.5867 | 0.00 | endometrium(1) | |
| 214780657 | MS | R406Q | 0.6028 | 0.7262 | rs587780014 | 0.0000412 | endometrium(1) |
| 214730490 | MS | R641Q | 0.2660 | 0.6668 | rs752870879 | 0.0000082 | endometrium(1) |
| 214781449 | SG | S142 * | 0.1272 | 0.00 | endometrium(1) | ||
| 214745097 | MS | L625I | 0.5980 | 0.2316 | 0.00 | endometrium(1) | |
| 214780981 | MS | V298A | 0.4174 | 0.9235 | 0.00 | endometrium(1) | |
| 214728928 | SY | L694L | rs139620052 | 0.000157 | endometrium(1) | ||
| 214745741 | SY | Y597Y | 0.00 | endometrium(1) | |||
| 214767501 | MS | G517R | 0.2520 | 0.0149 | 0.00 | hematopoietic_and_lymphoid_tissue(1) | |
| 214767558 | MS | L498I | 0.1770 | 0.0629 | 0.00 | ||
| 214730463 | MS | K650T | 0.3204 | 0.2411 | 0.00 | kidney(1) | |
| 214767483 | MS | V523I | 0.2358 | 0.4735 | 0.00 | kidney(1) | |
| 214767560 | MS | P497L | 0.0730 | 0.0046 | 0.00 | kidney(1) | |
| 214781275 | MS | A200V | 0.3100 | 0.7394 | 0.00 | kidney(1) | |
| 214792349 | MS | M104I | 0.1630 | 0.0450 | rs752133770 | 0.000008 | kidney(1) |
| 214728757 | SY | R751R | rs750001065 | 0.00 | large_intestine(1) | ||
| 214728786 | MS | L742V | 0.4234 | 0.7816 | 0.00 | large_intestine(1) | |
| 214728839 | MS | A724V | 0.0356 | 0.0153 | rs587782662 | 0.000041 | large_intestine(1) |
| 214728971 | SG | W680 * | 0.0967 | 0.00 | large_intestine(1) | ||
| 214745779 | MS | L585F | 0.2630 | 0.0431 | 0.00 | large_intestine(1) | |
| 214752448 | MS | V559A | 0.0894 | 0.2909 | 0.00 | large_intestine(1) | |
| 214767651 | MS | E467K | 0.1858 | 0.0168 | 0.00 | large_intestine(1) | |
| 214780632 | MS | M414I | 0.7050 | 0.5045 | 0.00 | large_intestine(1) | |
| 214780885 | MS | S330I | 0.6160 | 0.6216 | 0.00 | large_intestine(1) | |
| 214780904 | MS | H324N | 0.6444 | 0.6244 | 0.00 | large_intestine(1) | |
| 214780960 | MS | K305T | 0.7818 | 0.5088 | 0.00 | large_intestine(1) | |
| 214780974 | SY | P300P | 0.00 | large_intestine(1) | |||
| 214781327 | MS | V183L | 0.5944 | 0.3666 | 0.00 | large_intestine(1) | |
| 214781495 | MS | K127E | 0.7706 | 0.7394 | 0.00 | ||
| 214792327 | SG | R112 * | 0.0323 | rs758972589 | 0.000008 | large_intestine(1) | |
| 214752523 | MS | T534K | 0.3864 | 0.0564 | 0.00 | ||
| 214752535 | MS | P530L | 0.0214 | 0.0078 | 0.00 | liver(1) | |
| 214781157 | SY | L239L | rs760951875 | 0.000009 | |||
| 214781422 | MS | S151N | 0.3204 | 0.1398 | 0.00 | liver(1) | |
| 214792355 | MS | D102E | 0.5138 | 0.1463 | 0.00 | ||
| 214809564 | SY | P2P | 0.00 | liver(1) | |||
| 214728989 | MS | G674V | 0.1270 | 0.0055 | 0.00 | lung(1) | |
| 214730432 | MS | S660R | 0.2322 | 0.0874 | 0.00 | lung(1) | |
| 214745727 | MS | S602I | 0.6860 | 0.1905 | 0.00 | lung(1) | |
| 214745805 | MS | G576V | 0.4174 | 0.1383 | 0.00 | lung(1) | |
| 214752450 | SY | S558S | 0.00 | lung(1) | |||
| 214767507 | MS | S515A | 0.5474 | 0.5937 | 0.00 | lung(1) | |
| 214767582 | MS | T490P | 0.4354 | 0.5565 | 0.00 | lung(1) | |
| 214780637 | MS | A413T | 0.3100 | 0.8492 | 0.00 | lung(1) | |
| 214780642 | MS | P411H | 0.3420 | 0.0515 | 0.00 | lung(1) | |
| 214780818 | SY | V352V | rs768469265 | 0.000008 | lung(1) | ||
| 214780842 | MS | S344R | 0.6530 | 0.5045 | 0.00 | lung(1) | |
| 214780856 | MS | I340F | 0.8810 | 0.8826 | 0.00 | ||
| 214781201 | MS | E225K | 0.2884 | 0.6445 | 0.00 | ||
| 214781353 | MS | S174I | 0.7774 | 0.7572 | 0.00 | lung(1) | |
| 214781387 | MS | V163M | 0.2284 | 0.3354 | 0.00 | lung(1) | |
| 214797074 | MS | H68Y | 0.0190 | 0.0139 | 0.00 | ||
| 214809509 | MS | R21C | 0.2122 | 0.7193 | 0.00 | lung(1) | |
| 214769252 | MS | H459Y | 0.2160 | 0.2473 | 0.00 | NS(1) | |
| 214769253 | SY | D458D | 0.00 | NS(1) | |||
| 214769270 | MS | D453N | 0.2630 | 0.1696 | 0.00 | NS(1) | |
| 214809491 | MS | E27K | 0.1308 | 0.5288 | 0.00 | NS(1) | |
| 214728731 | MS | S760L | 0.2122 | 0.0823 | rs730881425 | 0.000025 | esophagus(1) |
| 214728819 | MS | R731C | 0.2852 | 0.3666 | rs76744638 | 0.000041 | esophagus(1) |
| 214745127 | MS | Q615E | 0.4234 | 0.1915 | rs751710099 | 0.000008 | esophagus(1) |
| 214767505 | SY | S515S | 0.00 | esophagus(1) | |||
| 214767573 | MS | Q493E | 0.5370 | 0.5719 | 0.00 | esophagus(1) | |
| 214780709 | MS | S389R | 0.5944 | 0.4714 | 0.00 | esophagus(1) | |
| 214781120 | MS | P252S | 0.3204 | 0.8220 | rs758368819 | 0.000009 | esophagus(1) |
| 214781351 | MS | A175P | 0.8810 | 0.4547 | 0.00 | esophagus(1) | |
| 214745744 | SY | K596K | rs777084777 | 0.000066 | pancreas(1) | ||
| 214752510 | SY | S538S | rs781448650 | 0.000016 | pancreas(1) | ||
| 214767635 | MS | G472E | 0.1042 | 0.0035 | 0.00 | pancreas(1) | |
| 214792385 | MS | I92M | 0.4354 | 0.2288 | 0.00 | pancreas(1) | |
| 214780909 | MS | R322H | 0.6340 | 0.7262 | rs774251286 | 0.000017 | pituitary(1) |
| 214730417 | SY | E665E | 0.00 | prostate(1) | |||
| 214752536 | MS | P530S | 0.0144 | 0.0102 | rs760144724 | 0.000008 | prostate(1) |
| 214769261 | MS | V456I | 0.1904 | 0.4630 | 0.00 | prostate(1) | |
| 214781033 | MS | P281S | 0.1358 | 0.6871 | rs200059956 | 0.000017 | prostate(1) |
| 214781127 | SY | I249I | rs746551077 | 0.000009 | prostate(1) | ||
| 214781355 | SY | A173A | 0.00 | prostate(1) | |||
| 214728723 | MS | F763V | 0.6444 | 0.3044 | rs761586960 | 0.000008 | skin(1) |
| 214730442 | MS | P657L | 0.0304 | 0.0144 | 0.00 | skin(1) | |
| 214730443 | MS | P657S | 0.1270 | 0.1673 | 0.00 | ||
| 214745129 | MS | V614A | 0.1114 | 0.5492 | 0.00 | skin(1) | |
| 214745834 | SY | R566R | 0.00 | skin(1) | |||
| 214767584 | MS | T489I | 0.1730 | 0.2288 | 0.00 | skin(1) | |
| 214767639 | MS | H471Y | 0.2284 | 0.1872 | rs867587389 | 0.00 | skin(1) |
| 214769288 | MS | L447F | 0.1204 | 0.0426 | 0.00 | skin(1) | |
| 214780570 | MS | A435V | 0.3420 | 0.0168 | 0.00 | skin(1) | |
| 214780609 | MS | V422G | 0.8136 | 0.8197 | rs76824305 | 0.000232 | skin(1) |
| 214780643 | MS | P411S | 0.0962 | 0.1509 | 0.00 | skin(1) | |
| 214780882 | MS | P331L | 0.3898 | 0.2518 | 0.00 | skin(1) | |
| 214781320 | MS | P185L | 0.4934 | 0.8492 | 0.00 | skin(1) | |
| 214781323 | MS | S184F | 0.1858 | 0.2233 | rs184660818 | 0.00 | skin(1) |
| 214792395 | MS | P89Q | 0.1056 | 0.0249 | 0.00 | skin(1) | |
| 214792396 | MS | P89S | 0.0212 | 0.0417 | 0.00 | skin(1) | |
| 214809508 | MS | R21L | 0.2038 | 0.6894 | 0.00 | skin(1) | |
| 214728752 | MS | G753D | 0.0762 | 0.0168 | rs867281641 | 0.00 | stomach(1) |
| 214728780 | MS | N744D | 0.5416 | 0.4141 | 0.00 | stomach(1) | |
| 214728831 | MS | D727N | 0.3044 | 0.5867 | rs730881424 | 0.000025 | stomach(1) |
| 214728891 | MS | P707S | 0.0436 | 0.0289 | 0.00 | stomach(1) | |
| 214728985 | SY | C675C | 0.00 | stomach(1) | |||
| 214730411 | SY | L667L | 0.00 | stomach(1) | |||
| 214767511 | SY | L513L | 0.00 | stomach(1) | |||
| 214767611 | MS | L480S | 0.1242 | 0.0086 | rs149839922 | 0.000008 | stomach(1) |
| 214769254 | MS | D458V | 0.0432 | 0.0034 | 0.00 | stomach(1) | |
| 214780751 | MS | T375A | 0.5088 | 0.8954 | 0.00 | stomach(1) | |
| 214781250 | FI | K208KENFS * | 0.0618 | rs587780033 | 0.000017 | stomach(1) | |
| 214781418 | SY | K152K | 0.00 | stomach(1) | |||
| 214792416 | MS | G82V | 0.4234 | 0.2775 | 0.00 | stomach(1) | |
| 214745083 | MS | W629C | 0.1678 | 0.0025 | 0.00 | ||
| 214781258 | MS | Q206K | 0.4174 | 0.6353 | 0.00 | ||
| 214728689 | MS | P774L | 0.3692 | 0.1073 | 0.00 | upper_aerodigestive_tract(1) | |
| 214745730 | MS | D601G | 0.6122 | 0.1612 | rs767765131 | 0.000016 | |
| 214769271 | MS | S452R | 0.5370 | 0.1476 | 0.00 | upper_aerodigestive_tract(1) | |
| 214781182 | MS | S231C | 0.5654 | 0.5912 | 0.00 | upper_aerodigestive_tract(1) | |
| 214809454 | MS | A39G | 0.5572 | 0.4215 | 0.00 | ||
| 214745154 | MS | H606D | 0.1552 | 0.0017 | 0.00 | urinary_tract(1) | |
| 214781269 | MS | S202C | 0.3982 | 0.4655 | 0.00 | urinary_tract(1) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimmino, F.; Formicola, D.; Capasso, M. Dualistic Role of BARD1 in Cancer. Genes 2017, 8, 375. https://doi.org/10.3390/genes8120375
Cimmino F, Formicola D, Capasso M. Dualistic Role of BARD1 in Cancer. Genes. 2017; 8(12):375. https://doi.org/10.3390/genes8120375
Chicago/Turabian StyleCimmino, Flora, Daniela Formicola, and Mario Capasso. 2017. "Dualistic Role of BARD1 in Cancer" Genes 8, no. 12: 375. https://doi.org/10.3390/genes8120375
APA StyleCimmino, F., Formicola, D., & Capasso, M. (2017). Dualistic Role of BARD1 in Cancer. Genes, 8(12), 375. https://doi.org/10.3390/genes8120375