A Microbiomic Analysis in African Americans with Colonic Lesions Reveals Streptococcus sp.VT162 as a Marker of Neoplastic Transformation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Preparation
2.3. Metabolomics Analysis
2.4. DNA Extraction
2.5. 16S rRNA Gene Profiling
2.6. HITChip Analysis
2.7. 454 Pyrosequencing 16S rRNA Gene Profiling from Tissue Samples
2.8. Metagenomic Analysis
2.9. Streptococcus sp. VT_162 Q-PCR
3. Results
3.1. Metabolomic Analysis of Adenoma Patients and Healthy Subjects’ Stool Samples
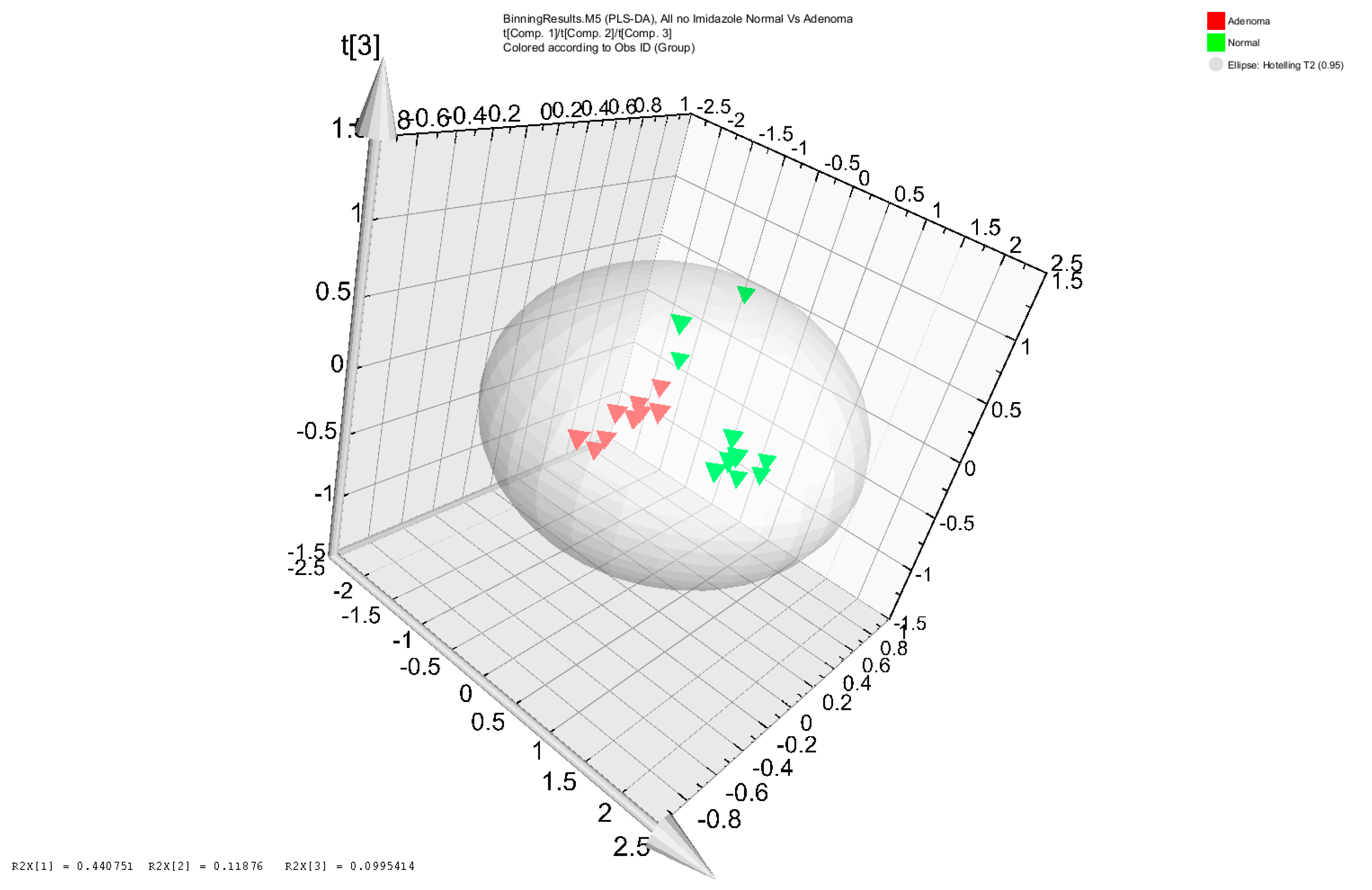
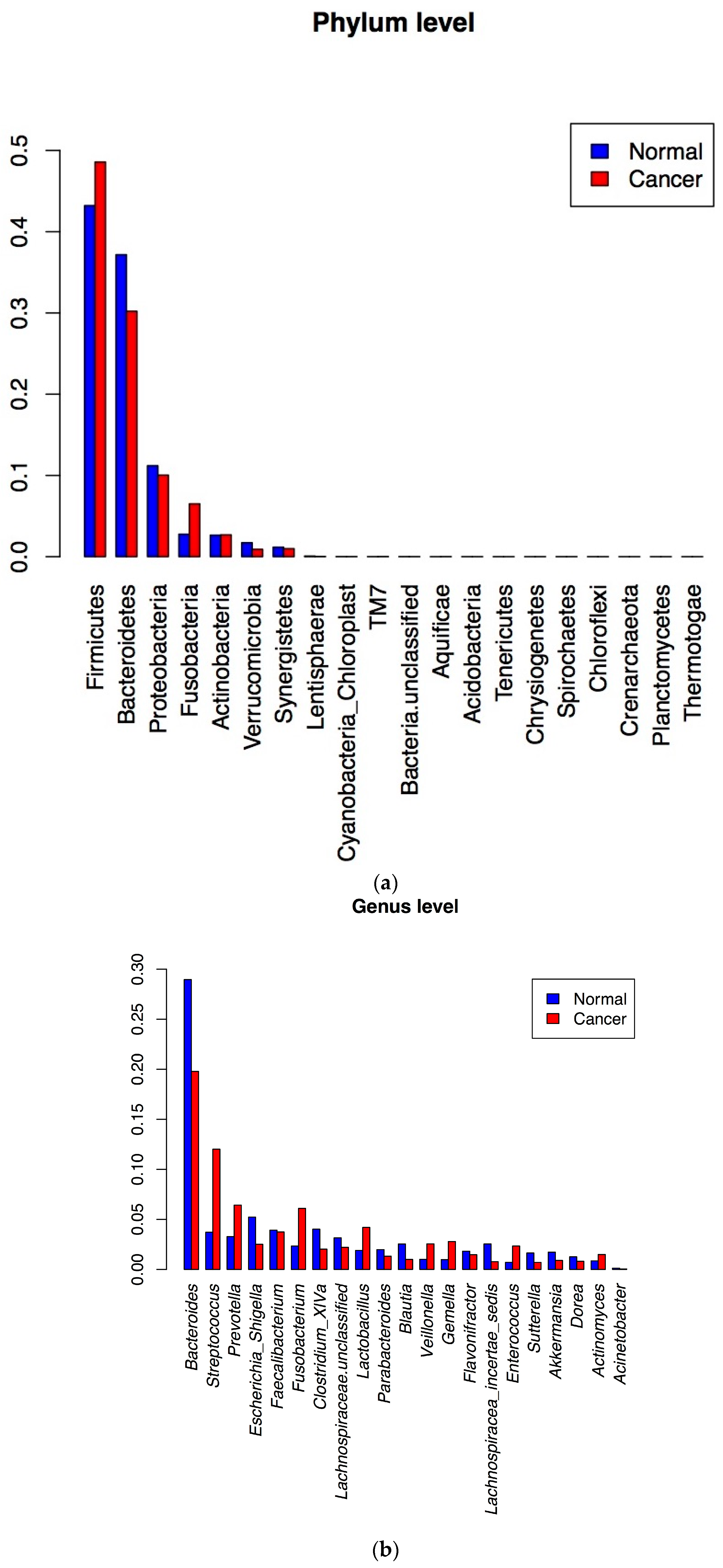
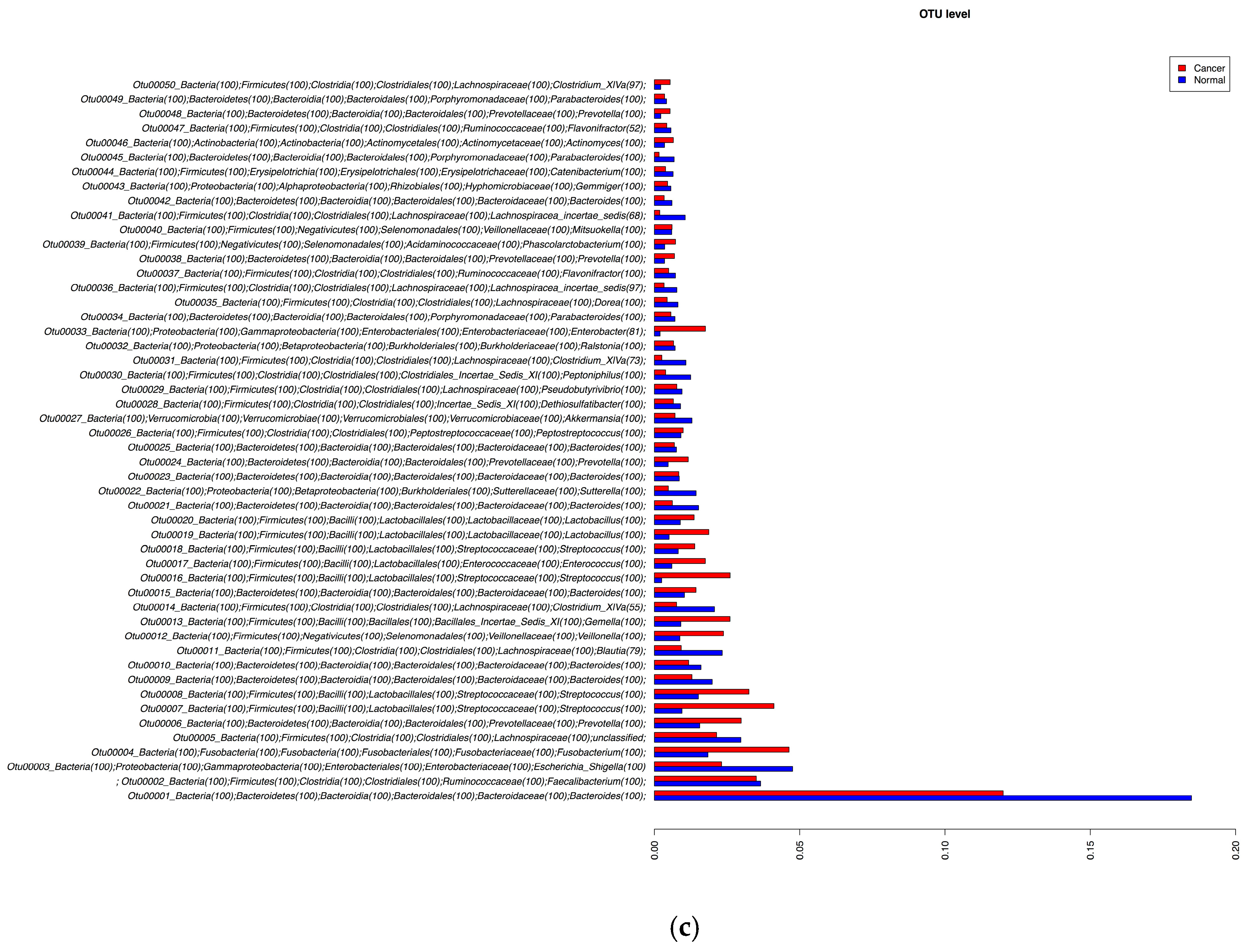
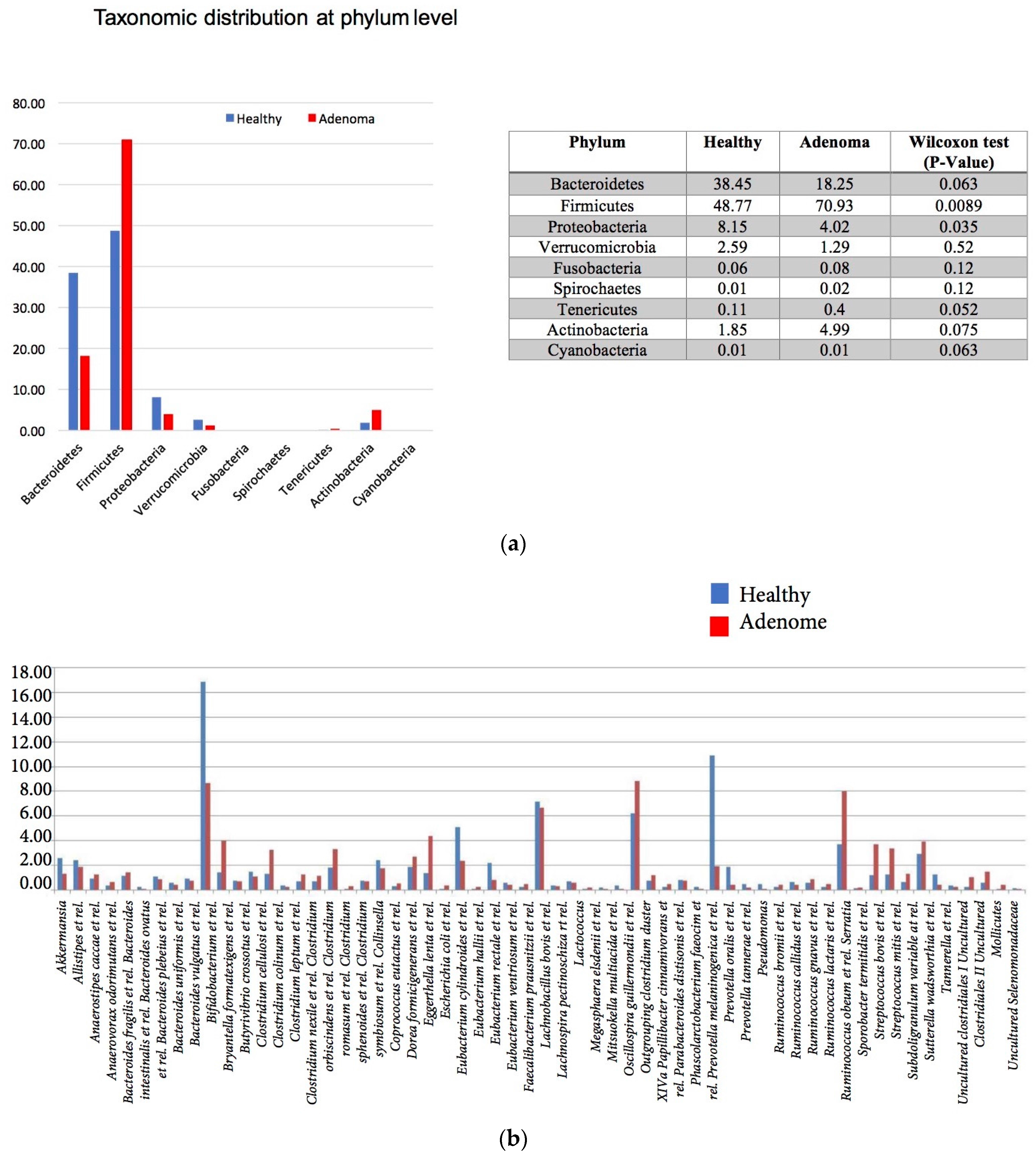
3.2. 16S rRNA Gene Analysis
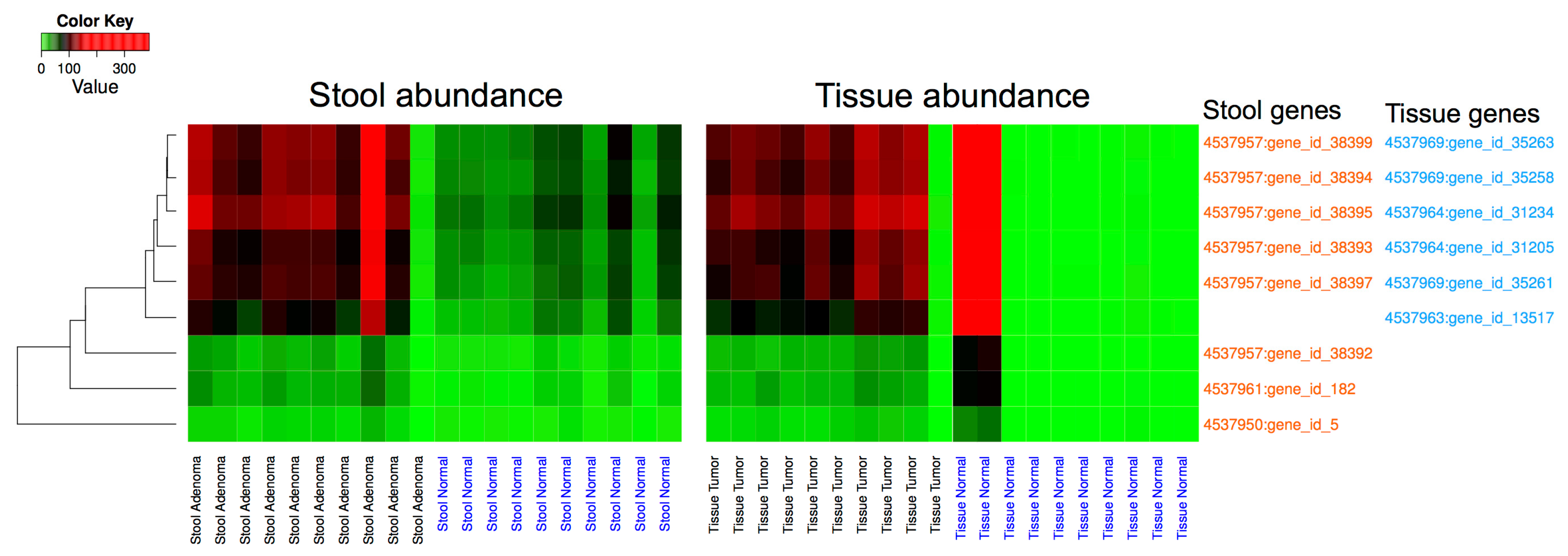
3.3. Metagenomic Data Analysis
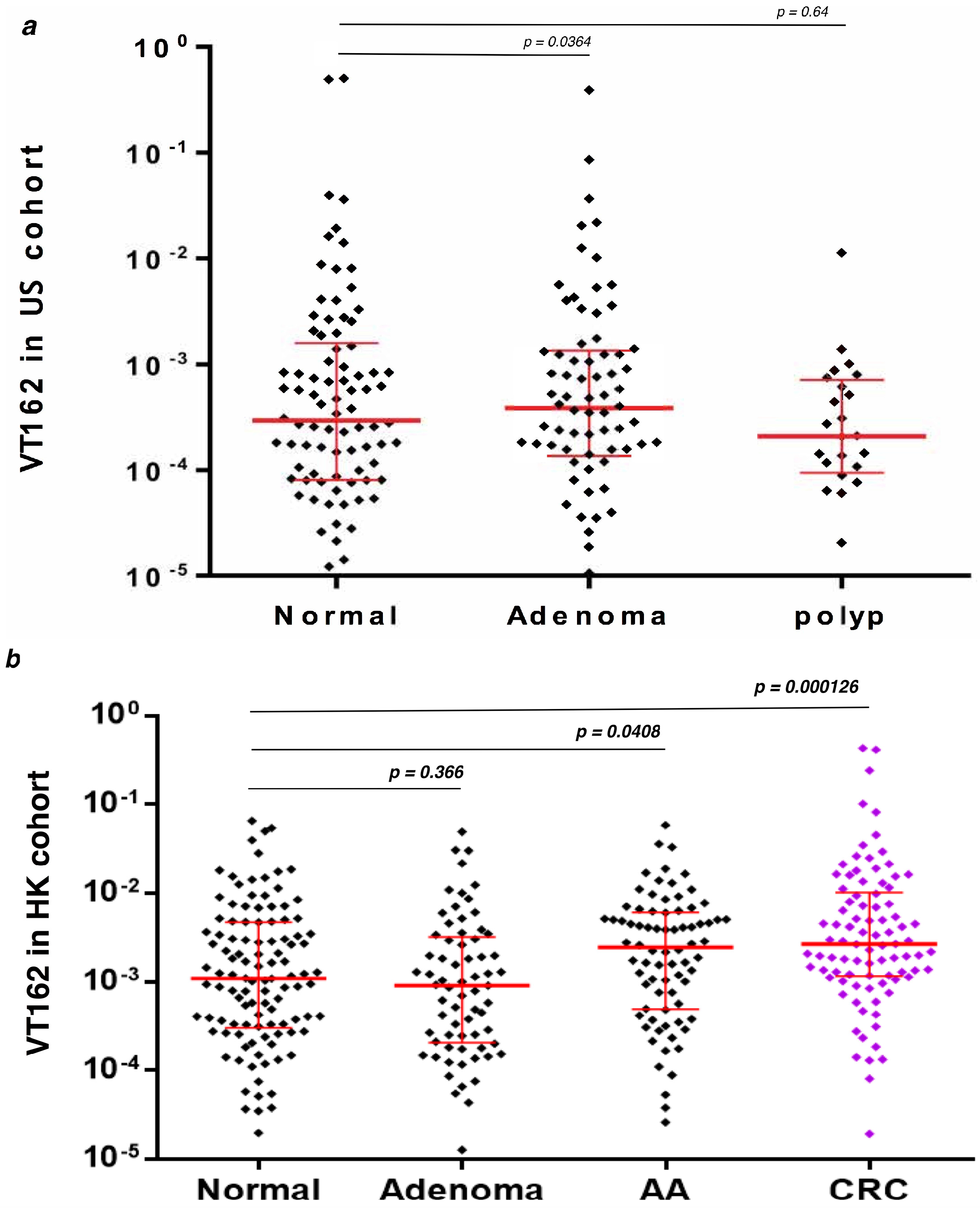
3.4. q-PCR Detection of Streptococcus sp VT_162 in Two Validation Cohorts
4. Discussion
Supplementary Materials
Acknowledgments
Authors Contribution
Conflicts of Interest
References
- Sinicrope, P.S.; Goode, E.L.; Limburg, P.J.; Vernon, S.W.; Wick, J.B.; Patten, C.A.; Decker, P.A.; Hanson, A.C.; Smith, C.M.; Beebe, T.J.; et al. A population-based study of prevalence and adherence trends in average risk colorectal cancer screening, 1997 to 2008. Cancer Epidemiol. Biomark. Prev. 2012, 21, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Friedenberg, F.K.; Singh, M.; George, N.S.; Sankineni, A.; Shah, S. Prevalence and distribution of adenomas in black Americans undergoing colorectal cancer screening. Dig. Dis. Sci. 2012, 57, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Blumenstein, I.; Tacke, W.; Bock, H.; Filmann, N.; Lieber, E.; Zeuzem, S.; Trojan, J.; Herrmann, E.; Schroder, O. Prevalence of colorectal cancer and its precursor lesions in symptomatic and asymptomatic patients undergoing total colonoscopy: Results of a large prospective, multicenter, controlled endoscopy study. Eur. J. Gastroenterol. Hepatol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.L.; Persky, V.; Turyk, M. Carcinoma in situ of the colorectum: SEER trends by race, gender, and total colorectal cancer. J. Surg. Oncol. 1999, 71, 123–129. [Google Scholar] [CrossRef]
- Smith, R.A.; von Eschenbach, A.C.; Wender, R.; Levin, B.; Byers, T.; Rothenberger, D.; Brooks, D.; Creasman, W.; Cohen, C.; Runowicz, C.; et al. American Cancer Society guidelines for the early detection of cancer: Update of early detection guidelines for prostate, colorectal, and endometrial cancers. Also: Update 2001—Testing for early lung cancer detection. CA Cancer J. Clin. 2001, 51, 38–75, quiz 77–80. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Bhupinderjit, A.; Bhutani, M.S.; Boardman, L.; Nguyen, C.; Romero, Y.; Srinivasan, R.; Figueroa-Moseley, C. Colorectal cancer in African Americans. Am. J. Gastroenterol. 2005, 100, 515–523, discussion 514. [Google Scholar] [CrossRef] [PubMed]
- Nouraie, M.; Hosseinkhah, F.; Brim, H.; Zamanifekri, B.; Smoot, D.T.; Ashktorab, H. Clinicopathological features of colon polyps from African-Americans. Dig. Dis. Sci. 2010, 55, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.L.; Dashwood, R.H.; Smoot, D. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig. Dis. Sci. 2009, 54, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Green, W.; Finzi, G.; Sessa, F.; Nouraie, M.; Lee, E.L.; Morgano, A.; Moschetta, A.; Cattaneo, M.; Mariani-Costantini, R.; et al. SEL1L, an UPR response protein, a potential marker of colonic cell transformation. Dig. Dis. Sci. 2012, 57, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Nguza, B.; Fatemi, M.; Nouraie, M.; Smoot, D.T.; Schaffer, A.A.; Kupfer, S.S.; Camargo, C.A., Jr.; Brim, H. Case-control study of vitamin D, dickkopf homolog 1 (DKK1) gene methylation, VDR gene polymorphism and the risk of colon adenoma in African Americans. PLoS ONE 2011, 6, e25314. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Schaffer, A.A.; Daremipouran, M.; Smoot, D.T.; Lee, E.; Brim, H. Distinct genetic alterations in colorectal cancer. PLoS ONE 2010, 5, e8879. [Google Scholar] [CrossRef] [PubMed]
- Brim, H.; Kumar, K.; Nazarian, J.; Hathout, Y.; Jafarian, A.; Lee, E.; Green, W.; Smoot, D.; Park, J.; Nouraie, M.; et al. SLC5A8 gene, a transporter of butyrate: A gut flora metabolite, is frequently methylated in African American colon adenomas. PLoS ONE 2011, 6, e20216. [Google Scholar] [CrossRef] [PubMed]
- Brim, H.; Lee, E.; Abu-Asab, M.S.; Chaouchi, M.; Razjouyan, H.; Namin, H.; Goel, A.; Schaffer, A.A.; Ashktorab, H. Genomic aberrations in an African American colorectal cancer cohort reveals a MSI-specific profile and chromosome X amplification in male patients. PLoS ONE 2012, 7, e40392. [Google Scholar] [CrossRef] [PubMed]
- Brim, H.; Mokarram, P.; Naghibalhossaini, F.; Saberi-Firoozi, M.; Al-Mandhari, M.; Al-Mawaly, K.; Al-Mjeni, R.; Al-Sayegh, A.; Raeburn, S.; Lee, E.; et al. Impact of BRAF, MLH1 on the incidence of microsatellite instability high colorectal cancer in populations based study. Mol. Cancer 2008, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Brim, H.; Mokarram, P.; Naghibalhossaini, F.; Saberi-Firoozi, M.; Lee, E.; Giardiello, F.; Smoot, D.T.; Nouraie, M.; Ashktorab, H. Distinct BRAF (V600E) and KRAS mutations in high microsatellite instability sporadic colorectal cancer in African Americans. Clin. Cancer Res. 2009, 15, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L. A dynamic partnership: Celebrating our gut flora. Anaerobe 2005, 11, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Boado, Y.S.; Wilson, C.L.; Hooper, L.V.; Gordon, J.I.; Hultgren, S.J.; Parks, W.C. Bacterial exposure induces and activates matrilysin in mucosal epithelial cells. J. Cell Biol. 2000, 148, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Morin, P.J.; Maouyo, D.; Sears, C.L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 2003, 124, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.L.; Kallstrom, G.; Faith, J.J.; Reyes, A.; Moore, A.; Dantas, G.; Gordon, J.I. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 6252–6257. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Huycke, M.M.; Gaskins, H.R. Commensal bacteria, redox stress, and colorectal cancer: Mechanisms and models. Exp. Biol. Med. 2004, 229, 586–597. [Google Scholar] [CrossRef]
- Lax, A.J.; Thomas, W. How bacteria could cause cancer: One step at a time. Trends Microbiol. 2002, 10, 293–299. [Google Scholar] [CrossRef]
- McGarr, S.E.; Ridlon, J.M.; Hylemon, P.B. Diet, anaerobic bacterial metabolism, and colon cancer: A review of the literature. J. Clin. Gastroenterol. 2005, 39, 98–109. [Google Scholar] [PubMed]
- Zoetendal, E.G.; Rajilic-Stojanovic, M.; de Vos, W.M. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008, 57, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Balter, M. Taking stock of the human microbiome and disease. Science 2012, 336, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Rogers, M.A.; Ruffin, M.T.T.; Schloss, P.D. The Human Gut Microbiome as a Screening Tool for Colorectal Cancer. Cancer Prev. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.T.; Zackular, J.P.; Chen, G.Y.; Schloss, P.D. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Irrazabal, T.; Belcheva, A.; Girardin, S.E.; Martin, A.; Philpott, D.J. The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 2014, 54, 309–320. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Housseau, F.; Sears, C.L. Microbiota and immune responses in colon cancer: More to learn. Cancer J. 2014, 20, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Brim, H.; Yooseph, S.; Zoetendal, E.G.; Lee, E.; Torralbo, M.; Laiyemo, A.O.; Shokrani, B.; Nelson, K.; Ashktorab, H. Microbiome analysis of stool samples from African Americans with colon polyps. PLoS ONE 2013, 8, e81352. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.J.; Burgess, J.P.; Snyder, R.W.; Popp, J.A.; Fennell, T.R. Metabolomics of urine for the assessment of microvesicular lipid accumulation in the liver following isoniazid exposure. Metabolomics 2010, 6, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.; Snyder, R.; Burgess, J.; Myers, C.; Tyl, R.; Sloan, C.; Fennell, T. Metabolomics in the assessment of chemical-induced reproductive and developmental outcomes using non-invasive biological fluids: Application to the study of butylbenzyl phthalate. J. Appl. Toxicol. 2009, 29, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogert, B.; de Vos, W.M.; Zoetendal, E.G.; Kleerebezem, M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 2011, 77, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Rajilic-Stojanovic, M.; Heilig, H.G.; Molenaar, D.; Kajander, K.; Surakka, A.; Smidt, H.; de Vos, W.M. Development and application of the human intestinal tract chip, a phylogenetic microarray: Analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ. Microbiol. 2009, 11, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Jeraldo, P.; Chia, N.; Goldenfeld, N. On the suitability of short reads of 16S rRNA for phylogeny-based analyses in environmental surveys. Environ. Microbiol. 2011, 13, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.L.; Hugenholtz, P.; Keller, J.; Blackall, L.L. Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl. Environ. Microbiol. 1995, 61, 1910–1916. [Google Scholar] [PubMed]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Q.; Chiu, J.; Chen, Y.; Huang, Y.; Higashimori, A.; Fang, J.Y.; Brim, H.; Ashktorab, H.; Ng, S.C.; Ng, S.S.; et al. Fecal Bacteria Act as Novel Biomarkers for Non-Invasive Diagnosis of Colorectal Cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef]
- O’Keefe, S.J.; Ou, J.; Aufreiter, S.; O’Connor, D.; Sharma, S.; Sepulveda, J.; Fukuwatari, T.; Shibata, K.; Mawhinney, T. Products of the colonic microbiota mediate the effects of diet on colon cancer risk. J. Nutr. 2009, 139, 2044–2048. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.L.; Li, X.L.; Xi, P.B.; Zhang, J.; Wu, G.; Zhu, W.Y. Metabolism of select amino acids in bacteria from the pig small intestine. Amino Acids 2012, 42, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, J.; Yatabe, M.S.; Ishibashi, K.; Nozawa, Y.; Sanada, H. Early detection of colon cancer by amino acid profiling using AminoIndex Technology: A case report. Diagn. Pathol. 2013, 8, 203. [Google Scholar] [CrossRef] [PubMed]
- Magrone, T.; Jirillo, E. The interplay between the gut immune system and microbiota in health and disease: Nutraceutical intervention for restoring intestinal homeostasis. Curr. Pharm. Des. 2013, 19, 1329–1342. [Google Scholar] [PubMed]
- Ray, K. Colorectal cancer: Fusobacterium nucleatum found in colon cancer tissue—Could an infection cause colorectal cancer? Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 662. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.N.; Araujo-Perez, F.; Azcarate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Takamura, N.; Kenzaka, T.; Minami, K.; Matsumura, M. Infective endocarditis caused by Streptococcus gallolyticus subspecies pasteurianus and colon cancer. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Shelburne, S.A.; Sahasrabhojane, P.; Saldana, M.; Yao, H.; Su, X.; Horstmann, N.; Thompson, E.; Flores, A.R. Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerg. Infect. Dis. 2014, 20, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Corredoira, J.; Coira, A.; Iniguez, I.; Pita, J.; Varela, J.; Alonso, M.P. Advanced intestinal cancer associated with Streptococcus infantarius (former S. bovis II/1) sepsis. Int. J. Clin. Pract. 2013, 67, 1358–1359. [Google Scholar] [CrossRef] [PubMed]
- Galdy, S.; Nastasi, G. Streptococcus bovis endocarditis and colon cancer: Myth or reality? A case report and literature review. BMJ Case Rep. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogert, B.; Boekhorst, J.; Herrmann, R.; Smid, E.J.; Zoetendal, E.G.; Kleerebezem, M. Comparative genomics analysis of Streptococcus isolates from the human small intestine reveals their adaptation to a highly dynamic ecosystem. PLoS ONE 2013, 8, e83418. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.L.; Li, X.L.; Xi, P.B.; Zhang, J.; Wu, G.; Zhu, W.Y. L-Glutamine regulates amino acid utilization by intestinal bacteria. Amino Acids 2013, 45, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Shi, Y.; Zhou, X.; Wu, C.; Cao, P.; Xu, C.; Hou, D.; Wang, Y.; Zhou, L. Microbiota in the throat and risk factors of laryngeal carcinoma. Appl. Environ. Microbiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Vecherkovskaya, M.F.; Tetz, G.V.; Tetz, V.V. Complete Genome Sequence of the Streptococcus sp. Strain VT 162, Isolated from the Saliva of Pediatric Oncohematology Patients. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, Y.; Wang, J.; Guo, Y.; Zhang, Y.; Xiao, S. Detection of fusobacterium nucleatum and fadA adhesin gene in patients with orthodontic gingivitis and non-orthodontic periodontal inflammation. PLoS ONE 2014, 9, e85280. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brim, H.; Yooseph, S.; Lee, E.; Sherif, Z.A.; Abbas, M.; Laiyemo, A.O.; Varma, S.; Torralba, M.; Dowd, S.E.; Nelson, K.E.; et al. A Microbiomic Analysis in African Americans with Colonic Lesions Reveals Streptococcus sp.VT162 as a Marker of Neoplastic Transformation. Genes 2017, 8, 314. https://doi.org/10.3390/genes8110314
Brim H, Yooseph S, Lee E, Sherif ZA, Abbas M, Laiyemo AO, Varma S, Torralba M, Dowd SE, Nelson KE, et al. A Microbiomic Analysis in African Americans with Colonic Lesions Reveals Streptococcus sp.VT162 as a Marker of Neoplastic Transformation. Genes. 2017; 8(11):314. https://doi.org/10.3390/genes8110314
Chicago/Turabian StyleBrim, Hassan, Shibu Yooseph, Edward Lee, Zaki A. Sherif, Muneer Abbas, Adeyinka O. Laiyemo, Sudhir Varma, Manolito Torralba, Scot E. Dowd, Karen E. Nelson, and et al. 2017. "A Microbiomic Analysis in African Americans with Colonic Lesions Reveals Streptococcus sp.VT162 as a Marker of Neoplastic Transformation" Genes 8, no. 11: 314. https://doi.org/10.3390/genes8110314
APA StyleBrim, H., Yooseph, S., Lee, E., Sherif, Z. A., Abbas, M., Laiyemo, A. O., Varma, S., Torralba, M., Dowd, S. E., Nelson, K. E., Pathmasiri, W., Sumner, S., De Vos, W., Liang, Q., Yu, J., Zoetendal, E., & Ashktorab, H. (2017). A Microbiomic Analysis in African Americans with Colonic Lesions Reveals Streptococcus sp.VT162 as a Marker of Neoplastic Transformation. Genes, 8(11), 314. https://doi.org/10.3390/genes8110314