
Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of the Pre-Inca and Inca Mummies
2.2. Description of the Renaissance Mummies from Naples
2.3. DNA Extraction and Avoidance of Contamination
2.4. 16S ribosomal RNA Gene High-Throughput Sequencing
2.5. 16S ribosomal RNA Gene Sequence Analyses
2.6. Shotgun Metagenome Sequence Processing
2.7. Taxonomic Assignments of Shotgun Metagenomic Data
2.8. Sequences Associated with Diet, Metabolic Profiles and Carbohydrate-Active Enzymes
2.9. Putative Resistome Characterization
3. Results
3.1. Characterization of the Mummies
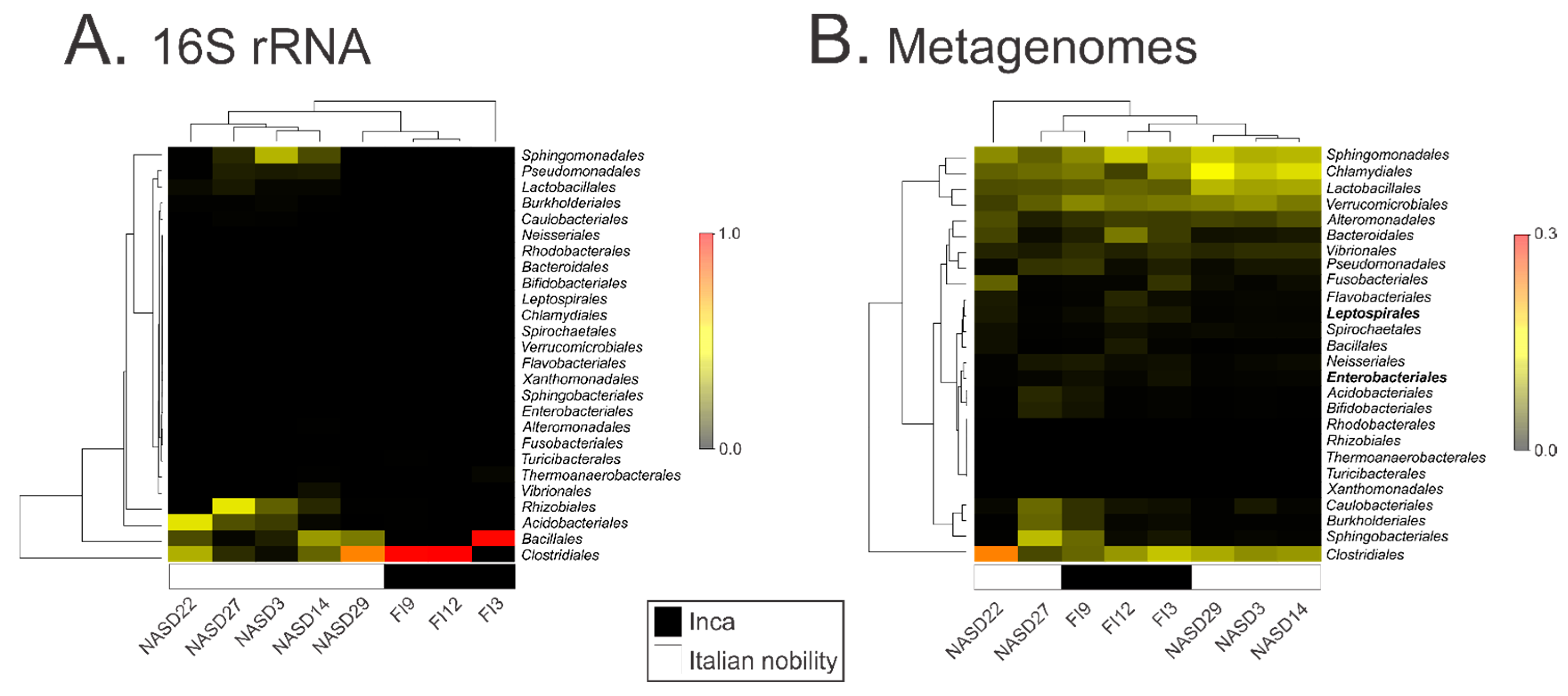
3.2. Characterization of the 16S ribosomal RNA Gene by High Throughput Sequencing
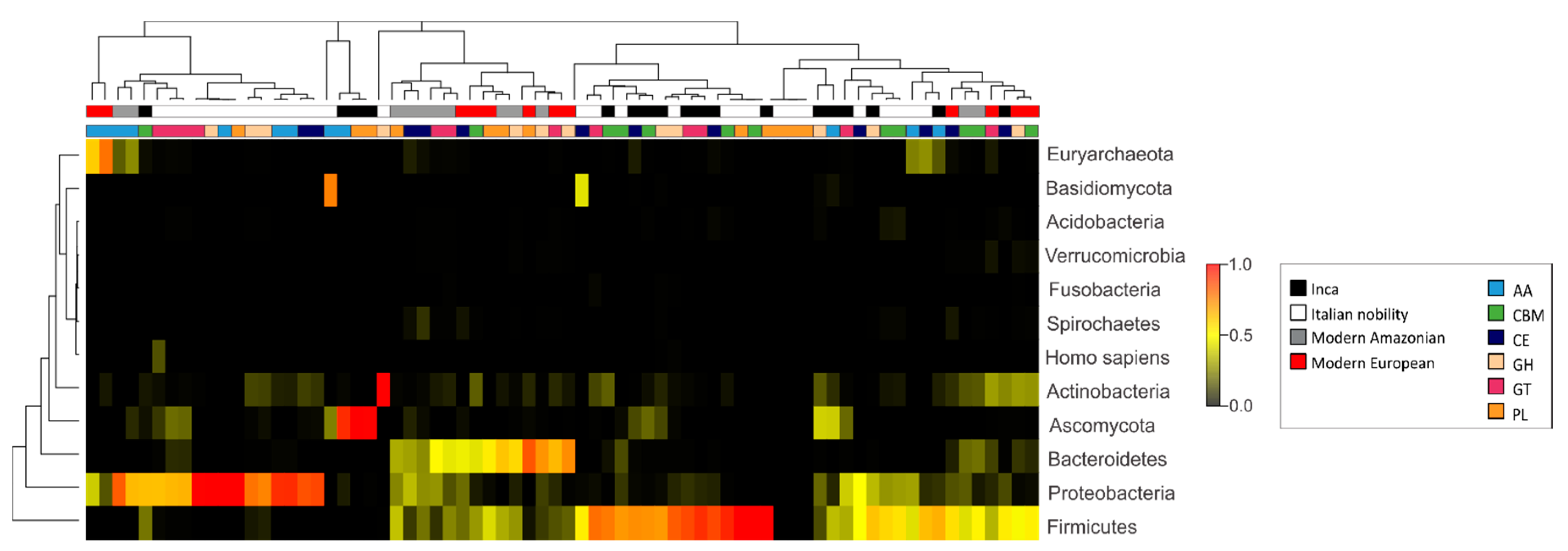
3.3. Characterization of the Gut Microbial Communities Using Shotgun Metagenomics
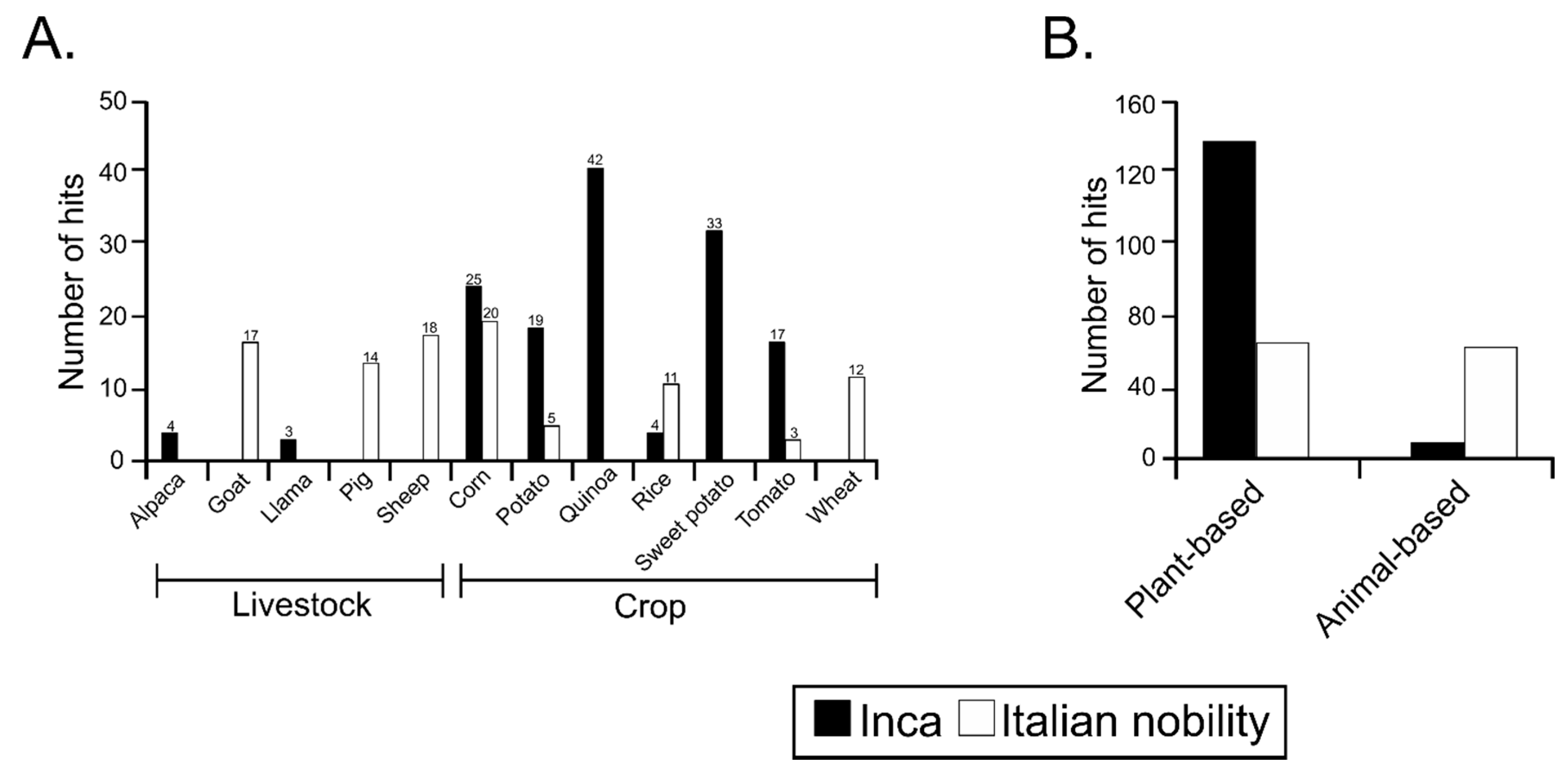
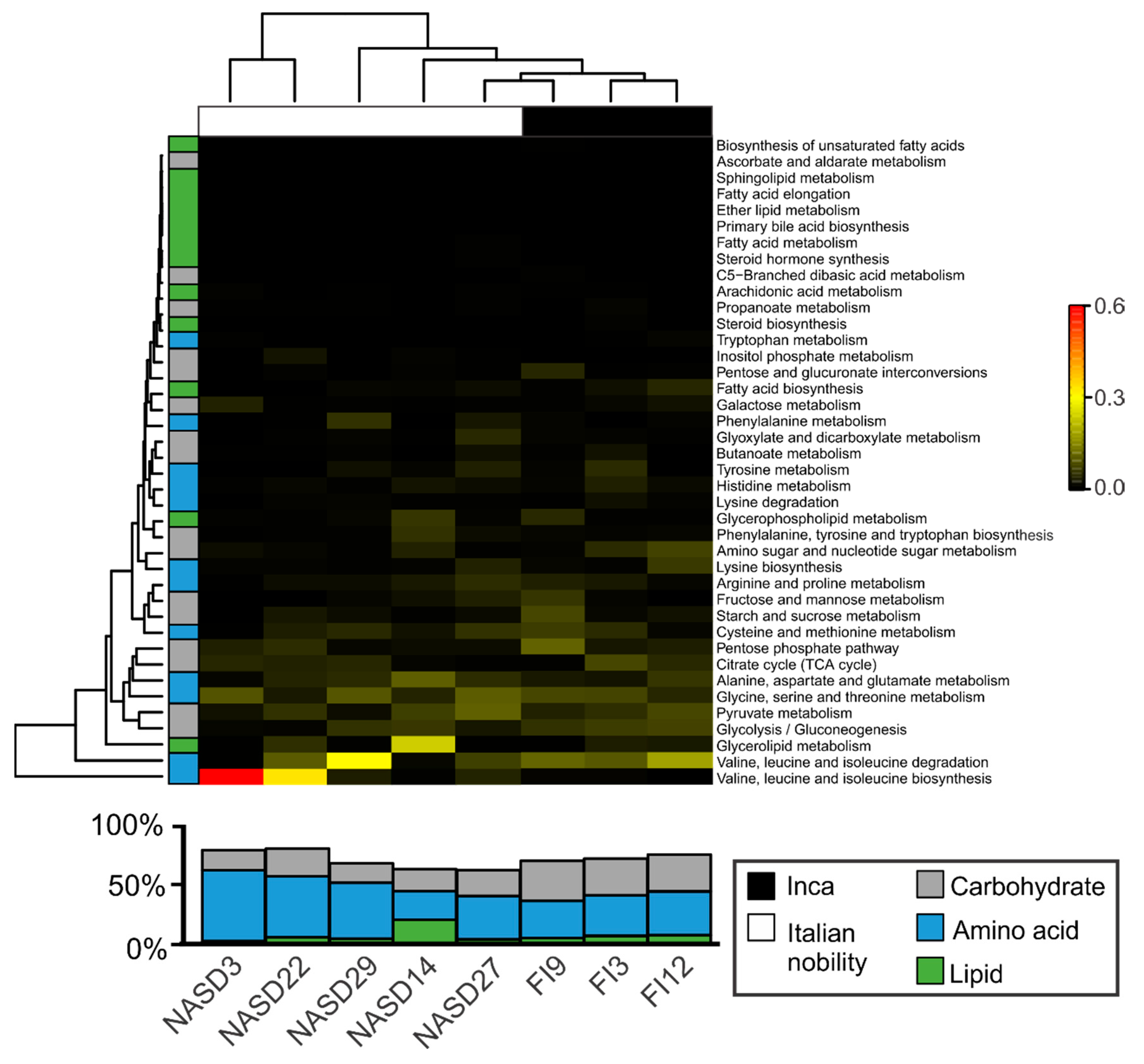
3.4. Sequences Associated with Dietary Habits and Metabolism
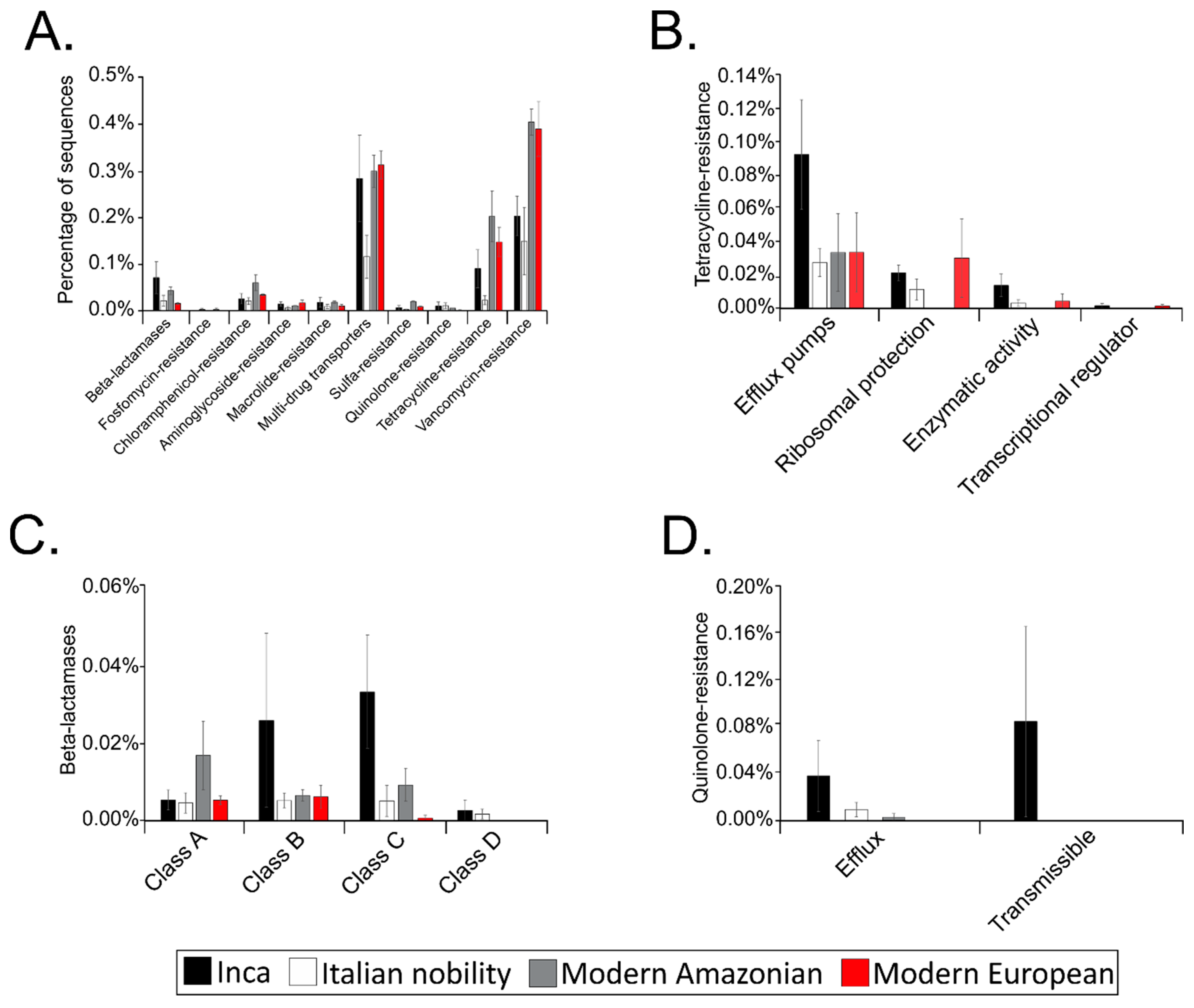
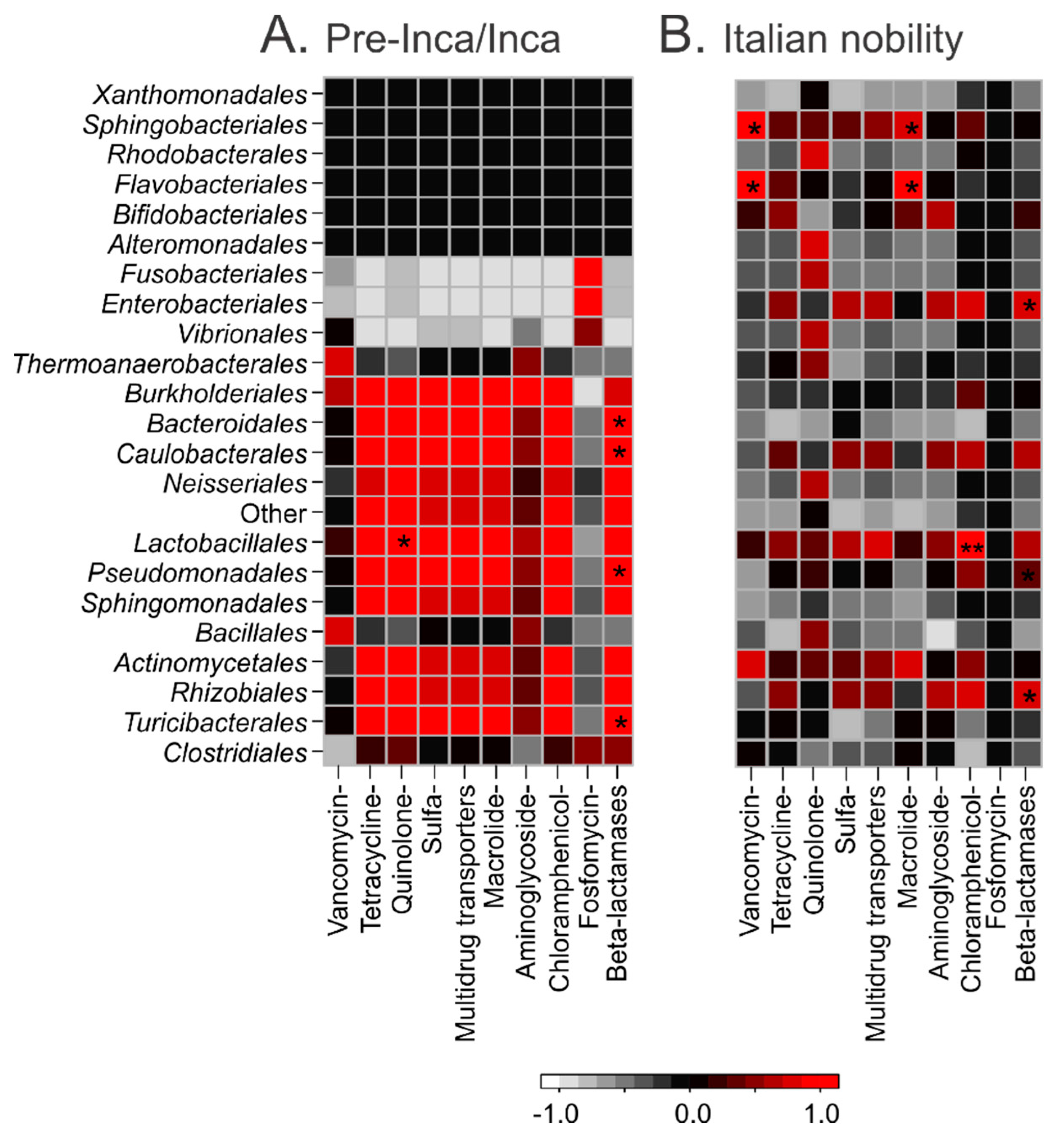
3.5. Putative Gut Resistomes of Pre-Inca/Inca and Italian Nobility Mummies
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Data Availability
References
- Adler, C.J.; Dobney, K.; Weyrich, L.S.; Kaidonis, J.; Walker, A.W.; Haak, W.; Bradshaw, C.J.; Townsend, G.; Soltysiak, A.; Alt, K.W.; et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 2013, 45, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P. Pulling teeth from history: DNA from ancient teeth can help to yield information about our ancestors’ health, diet and diseases. EMBO Rep. 2014, 15, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Speller, C.; Collins, M.J. A new era in palaeomicrobiology: Prospects for ancient dental calculus as a long-term record of the human oral microbiome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20130376. [Google Scholar] [CrossRef] [PubMed]
- Cano, R.J.; Rivera-Perez, J.; Toranzos, G.A.; Santiago-Rodriguez, T.M.; Narganes-Storde, Y.M.; Chanlatte-Baik, L.; Garcia-Roldan, E.; Bunkley-Williams, L.; Massey, S.E. Paleomicrobiology: Revealing fecal microbiomes of ancient indigenous cultures. PLoS ONE 2014, 9, e106833. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Narganes-Storde, Y.M.; Chanlatte, L.; Crespo-Torres, E.; Toranzos, G.A.; Jimenez-Flores, R.; Hamrick, A.; Cano, R.J. Microbial communities in pre-Columbian coprolites. PLoS ONE 2013, 8, e65191. [Google Scholar] [CrossRef] [PubMed]
- Appelt, S.; Armougom, F.; le Bailly, M.; Robert, C.; Drancourt, M. Polyphasic analysis of a middle ages coprolite microbiota, Belgium. PLoS ONE 2014, 9, e88376. [Google Scholar] [CrossRef] [PubMed]
- Appelt, S.; Fancello, L.; le Bailly, M.; Raoult, D.; Drancourt, M.; Desnues, C. Viruses in a 14th-century coprolite. Appl. Environ. Microbiol. 2014, 80, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Luciani, S.; Fornaciari, G.; Rickards, O.; Labarga, C.M.; Rollo, F. Molecular characterization of a pre-Columbian mummy and in situ coprolite. Am. J. Phys. Anthropol. 2006, 129, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Janaway, R.C.; Wilson, A.C.; Carpio-Díaz, G.; Guillen, S. Taphonomic Changes to the Buried Body in Arid Environments: An Experimental Case Study in Peru; Criminal and Environmental Soil Forensics; Springer: Dordrecht, The Netherland, 2009; pp. 341–356. [Google Scholar]
- Aufderheide, A.C. The Scientific Study of Mummies; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Vreeland, J.M. Mummies of Peru. In Mummies, Disease, and Ancient Cultures; Cambridge University Press: Cambridge, UK, 1998; pp. 154–189. [Google Scholar]
- Fornaciari, A.; Giuffra, V.; Pezzini, F. Secondary burial and mummification practices in the Kingdom of the two Sicilies. Mortality 2010, 15, 223–249. [Google Scholar] [CrossRef]
- Fornaciari, G. The Aragonese mummies of the Basilica of Saint domenico maggiore in Naples. Med. Secoli 2006, 18, 843–864. [Google Scholar] [PubMed]
- Ubaldi, M.; Luciani, S.; Marota, I.; Fornaciari, G.; Cano, R.J.; Rollo, F. Sequence analysis of bacterial DNA in the colon of an Andean mummy. Am. J. Phys. Anthropol. 1998, 107, 285–295. [Google Scholar] [CrossRef]
- Tito, R.Y.; Knights, D.; Metcalf, J.; Obregon-Tito, A.J.; Cleeland, L.; Najar, F.; Roe, B.; Reinhard, K.; Sobolik, K.; Belknap, S.; et al. Insights from characterizing extinct human gut microbiomes. PLoS ONE 2012, 7, e51146. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Gut Microbiome of an 11th Century AD Pre-Columbian Andean Mummy. PLoS ONE 2015, 10, e0138135. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Taxonomic and predicted metabolic profiles of the human gut microbiome in pre-Columbian mummies. FEMS Microbiol. Ecol. 2016, 92, fiw182. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol. Lett. 2016, 363, fnv219. [Google Scholar] [CrossRef] [PubMed]
- Piperno, D.R.; Dillehay, T.D. Starch grains on human teeth reveal early broad crop diet in northern Peru. Proc. Natl. Acad. Sci. USA 2008, 105, 19622–19627. [Google Scholar] [CrossRef] [PubMed]
- Livengood, S.V. Refining Dietary Estimates at Machu Picchu Using Combined Dental Macro/Microwear and Isotopic Analyses. Master’s Thesis, Georgia State University, Atlanta, GA, USA, 2012. [Google Scholar]
- Malpass, M.A. Daily Life in the Inca Empire; ABC-CLIO: Santa Barbara, CA, USA, 2009. [Google Scholar]
- Fornaciari, G. Food and disease at the Renaissance courts of Naples and Florence: A paleonutritional study. Appetite 2008, 51, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Riera-Melis, A. Society, food and Feudalism. In Food: A Culinary History from Antiquity to the Present; Flandrin, J.L., Montanari, M., Eds.; Columbia University: New York, NY, USA, 1999; pp. 251–267. [Google Scholar]
- Grieco, A.J. Food and social classes in late Medieval and Renaissance Italy. In Food: A Culinary History from Antiquity to the Present; Flandrin, J.L., Montanari, M., Eds.; Columbia University: New York, NY, USA, 1999; pp. 302–312. [Google Scholar]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Fornaciari, G.; Castagna, M.; Viacava, P.; Tognetti, A.; Bevilacqua, G.; Segura, E.L. Chagas’ disease in Peruvian Inca mummy. Lancet 1992, 339, 128–129. [Google Scholar] [CrossRef]
- Fornaciari, G. The mummies of the Abbey of Saint Domenico Maggiore in Naples: A preliminary report. Arch. Antrop. Etnol. 1985, 115, 21. [Google Scholar]
- Fornaciari, G.; Pollina, L.; Tornabuoni, D.; Tognetti, A. Pulmonary and hepatic pathologies in the series of mummies of S. Domenico Maggiore at Naples (XVI century). In Advances in Paleopathology: Proceeding of the VII European meeting of Paleopathology Association, Lyon, September 1988; M. Solfanelli: Chieti, Italy, 1988; pp. 107–115. [Google Scholar]
- Gaeta, R.; Ventura, L.; Fornaciari, G. Il tumore di Ferdinando Orsini, duca di Gravina di Puglia (+1549). In Atti del 50° Congrsso Nazionale della Società Italiana di Storia della Medicina; IRIS Università di Pisa: Palermo, Italy, 2015. [Google Scholar]
- Gaeta, R.; Ventura, L.; Fornaciari, G. The Cutaneous Cancer of Ferdinando Orsini, 5th Duke of Gravina. Jama Dermatol. 2017, 153, 643. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, R.; Giuffra, V.; Fornaciari, G. Cancer in the Renaissance court of Naples. Lancet Oncol. 2017, 18, e432. [Google Scholar] [CrossRef]
- Fornaciari, G. Histology of ancient soft tissue tumors: A review. Int. J. Paleopathol. 2017, in press. [Google Scholar]
- Marchetti, A.; Pellegrini, S.; Bevilacqua, G.; Fornaciari, G. K-RAS mutation in the tumour of Ferrante I of Aragon, King of Naples. Lancet 1996, 347, 1272. [Google Scholar] [CrossRef]
- Ottini, L.; Falchetti, M.; Marinozzi, S.; Angeletti, L.R.; Fornaciari, G. Gene-environment interactions in the pre–Industrial Era: The cancer of King Ferrante I of Aragon (1431–1494). Hum. Pathol. 2011, 42, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Ciranni, R.; Fornaciari, G. Juvenile cirrhosis in a 16th century Italian mummy. Current technologies in pathology and ancient human tissues. Virchows Archiv. 2004, 445, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, Y.; Qian, P.Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A.; et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [PubMed]
- Ginolhac, A.; Rasmussen, M.; Gilbert, M.T.; Willerslev, E.; Orlando, L. mapDamage: Testing for damage patterns in ancient DNA sequences. Bioinformatics 2011, 27, 2153–2155. [Google Scholar] [CrossRef] [PubMed]
- Pati, A.; Heath, L.S.; Kyrpides, N.C.; Ivanova, N. ClaMS: A Classifier for Metagenomic Sequences. Stand. Geno. Sci. 2011, 5, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Stearns, J.C.; Lynch, M.D.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef] [PubMed]
- Menguer, P.K.; Sperotto, R.A.; Ricachenevsky, F.K. A walk on the wild side: Oryza species as source for rice abiotic stress tolerance. Genet. Mol. Biol. 2017, 40, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Quesada, T.; Espinoza, A.M. Ultrastructure of the wild rice Oryza grandiglumis (Gramineae) in Costa Rica. Rev. Biol. Trop. 2006, 54, 377–385. [Google Scholar] [CrossRef] [PubMed]
- El Kaoutari, A.; Armougom, F.; Leroy, Q.; Vialettes, B.; Million, M.; Raoult, D.; Henrissat, B. Development and validation of a microarray for the investigation of the CAZymes encoded by the human gut microbiome. PLoS ONE 2013, 8, e84033. [Google Scholar] [CrossRef] [PubMed]
- Thaker, M.; Spanogiannopoulos, P.; Wright, G.D. The tetracycline resistome. Cell. Mol. Life Sci. 2010, 67, 419–431. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.H.; Jacoby, G.A. Mechanism of plasmid-mediated quinolone resistance. Proc. Natl. Acad. Sci. USA 2002, 99, 5638–5642. [Google Scholar] [CrossRef] [PubMed]
- Ziesemer, K.A.; Mann, A.E.; Sankaranarayanan, K.; Schroeder, H.; Ozga, A.T.; Brandt, B.W.; Zaura, E.; Waters-Rist, A.; Hoogland, M.; Salazar-Garcia, D.C.; et al. Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification. Sci. Rep. 2015, 5, 16498. [Google Scholar] [CrossRef] [PubMed]
- Maixner, F.; Thomma, A.; Cipollini, G.; Widder, S.; Rattei, T.; Zink, A. Metagenomic analysis reveals presence of Treponema denticola in a tissue biopsy of the Iceman. PLoS ONE 2014, 9, e99994. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Després, V.; Hebler, J.; Rohland, N.; Kuch, M.; Krause, J.; Vigilant, L.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hofstaedter, C.E.; Zhao, C.; Mattei, L.; Tanes, C.; Clarke, E.; Lauder, A.; Sherrill-Mix, S.; Chehoud, C.; Kelsen, J. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 2017, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- DeBruyn, J.M.; Hauther, K.A. Postmortem succession of gut microbial communities in human cadavers. PeerJ 2017, 5, e3437. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. npj Biofilms Microb. 2016, 2, 16004. [Google Scholar] [CrossRef] [PubMed]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, M.; Solyman, S.; Taha, M.; Hanora, A. Preliminary characterization of human skin microbiome in healthy Egyptian individuals. Cell Mol. Biol. 2016, 62, 21–27. [Google Scholar] [PubMed]
- Hamad, I.; Delaporte, E.; Raoult, D.; Bittar, F. Detection of termites and other insects consumed by African great apes using molecular fecal analysis. Sci. Rep. 2014, 4, 4478. [Google Scholar] [CrossRef] [PubMed]
- Nistelberger, H.M.; Smith, O.; Wales, N.; Star, B.; Boessenkool, S. The efficacy of high-throughput sequencing and target enrichment on charred archaeobotanical remains. Sci. Rep. 2016, 6, 37347. [Google Scholar] [CrossRef] [PubMed]
- Tasse, L.; Bercovici, J.; Pizzut-Serin, S.; Robe, P.; Tap, J.; Klopp, C.; Cantarel, B.L.; Coutinho, P.M.; Henrissat, B.; Leclerc, M. Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 2010, 20, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Turroni, S.; Severgnini, M.; Peano, C.; Brigidi, P.; Crittenden, A.N.; Henry, A.G.; Candela, M. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr. Biol. 2015, 25, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Sarkanen, S.; Wang, Y.Y. Lignin-degrading enzyme activities. Methods Mol. Biol. 2012, 908, 251–268. [Google Scholar] [PubMed]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013, 4, 2151. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Moe, L.A.; Rodbumrer, J.; Gaarder, A.; Handelsman, J. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J. 2009, 3, 243–251. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Toranzos, G.A.; Marota, I.; Giuffra, V.; Cano, R.J. Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies. Genes 2017, 8, 310. https://doi.org/10.3390/genes8110310
Santiago-Rodriguez TM, Fornaciari G, Luciani S, Toranzos GA, Marota I, Giuffra V, Cano RJ. Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies. Genes. 2017; 8(11):310. https://doi.org/10.3390/genes8110310
Chicago/Turabian StyleSantiago-Rodriguez, Tasha M., Gino Fornaciari, Stefania Luciani, Gary A. Toranzos, Isolina Marota, Valentina Giuffra, and Raul J. Cano. 2017. "Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies" Genes 8, no. 11: 310. https://doi.org/10.3390/genes8110310
APA StyleSantiago-Rodriguez, T. M., Fornaciari, G., Luciani, S., Toranzos, G. A., Marota, I., Giuffra, V., & Cano, R. J. (2017). Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies. Genes, 8(11), 310. https://doi.org/10.3390/genes8110310