
Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome †


Abstract
:1. Introduction
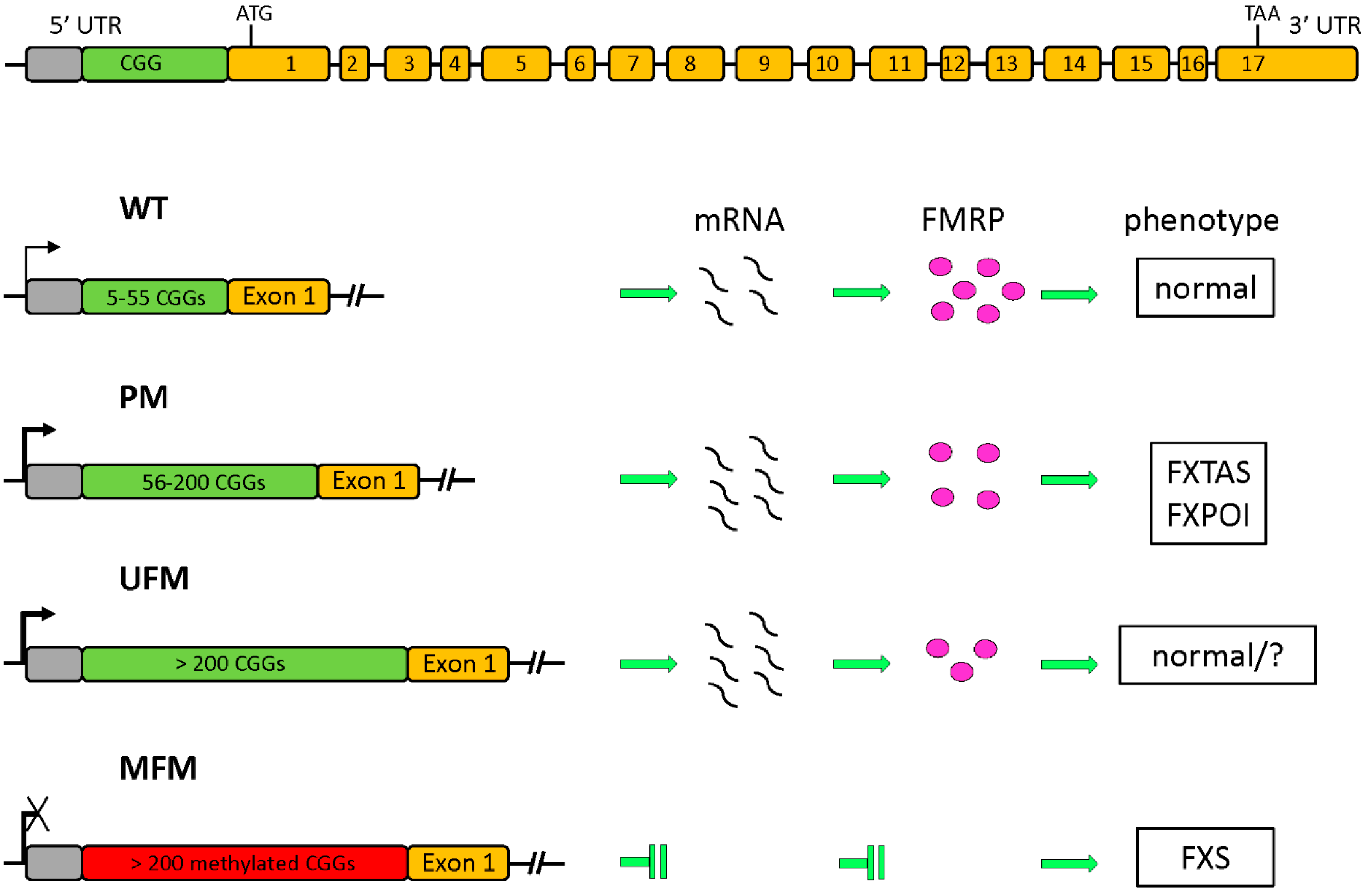
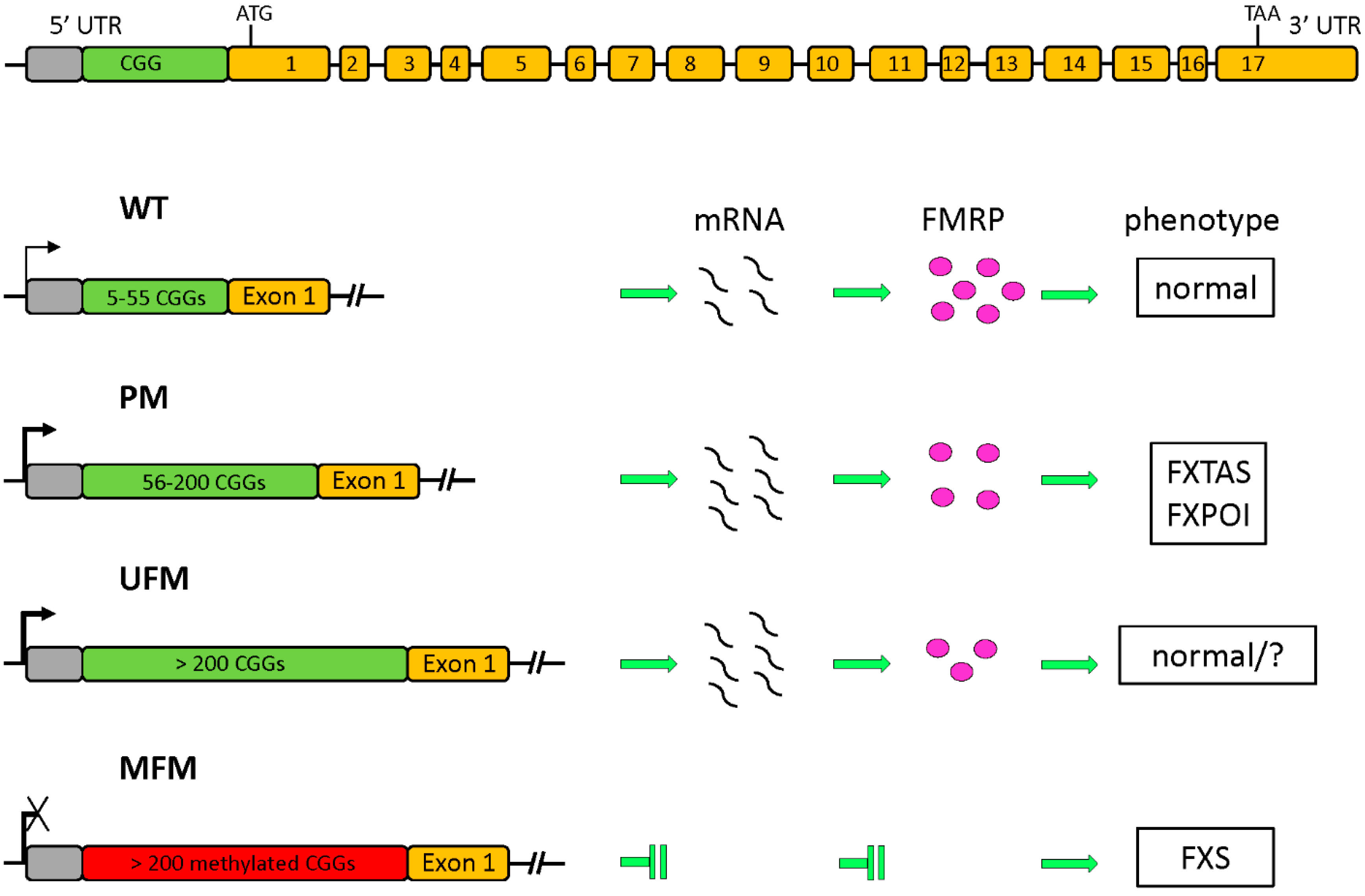
1.1. The FMR1 Gene and Its Protein Product
1.2. Epigenetic Regulation of FMR1 Transcription
2. Therapeutic Approaches for FXS
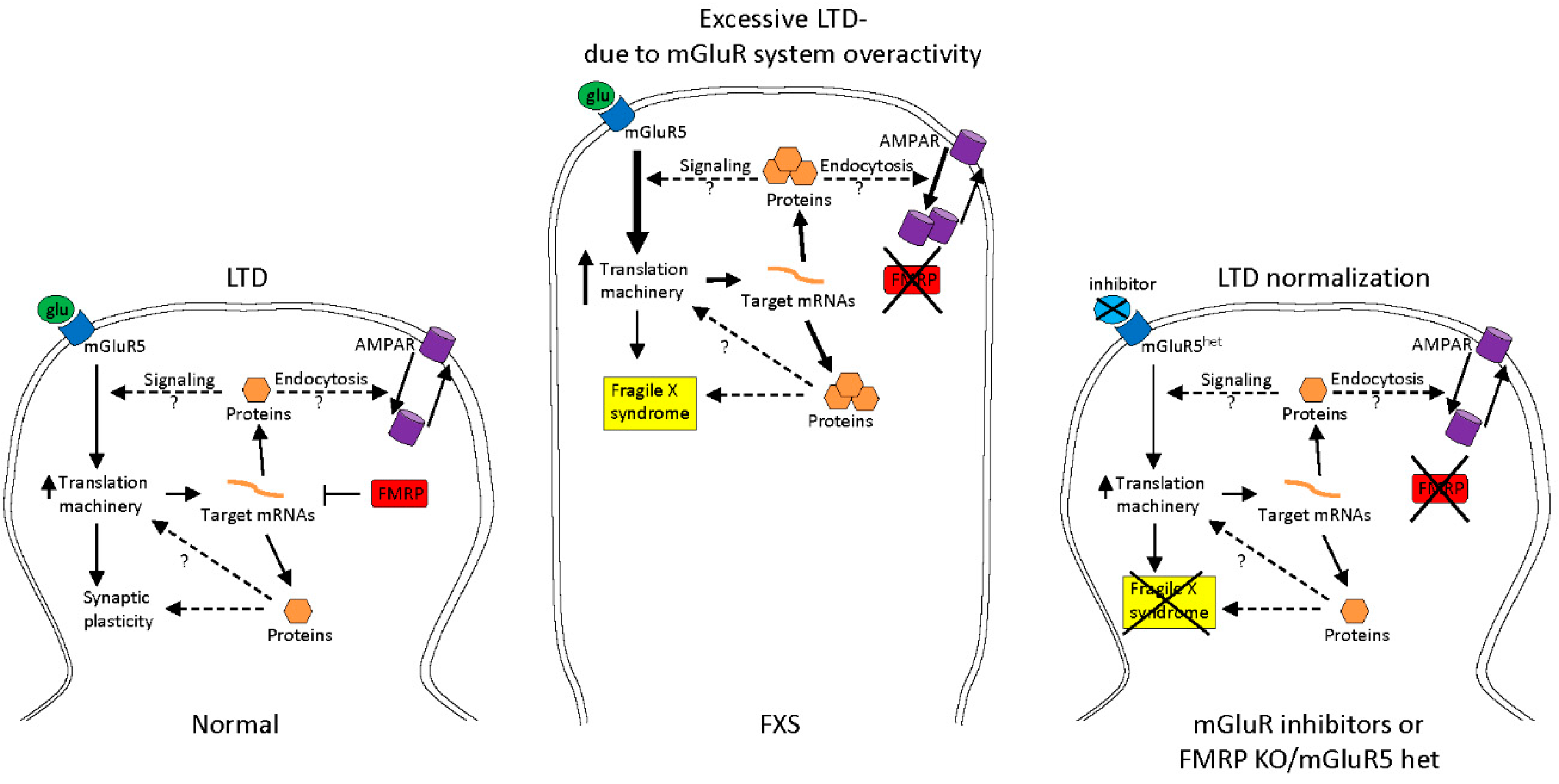
2.1. Treatments to Compensate for Lack of FMRP
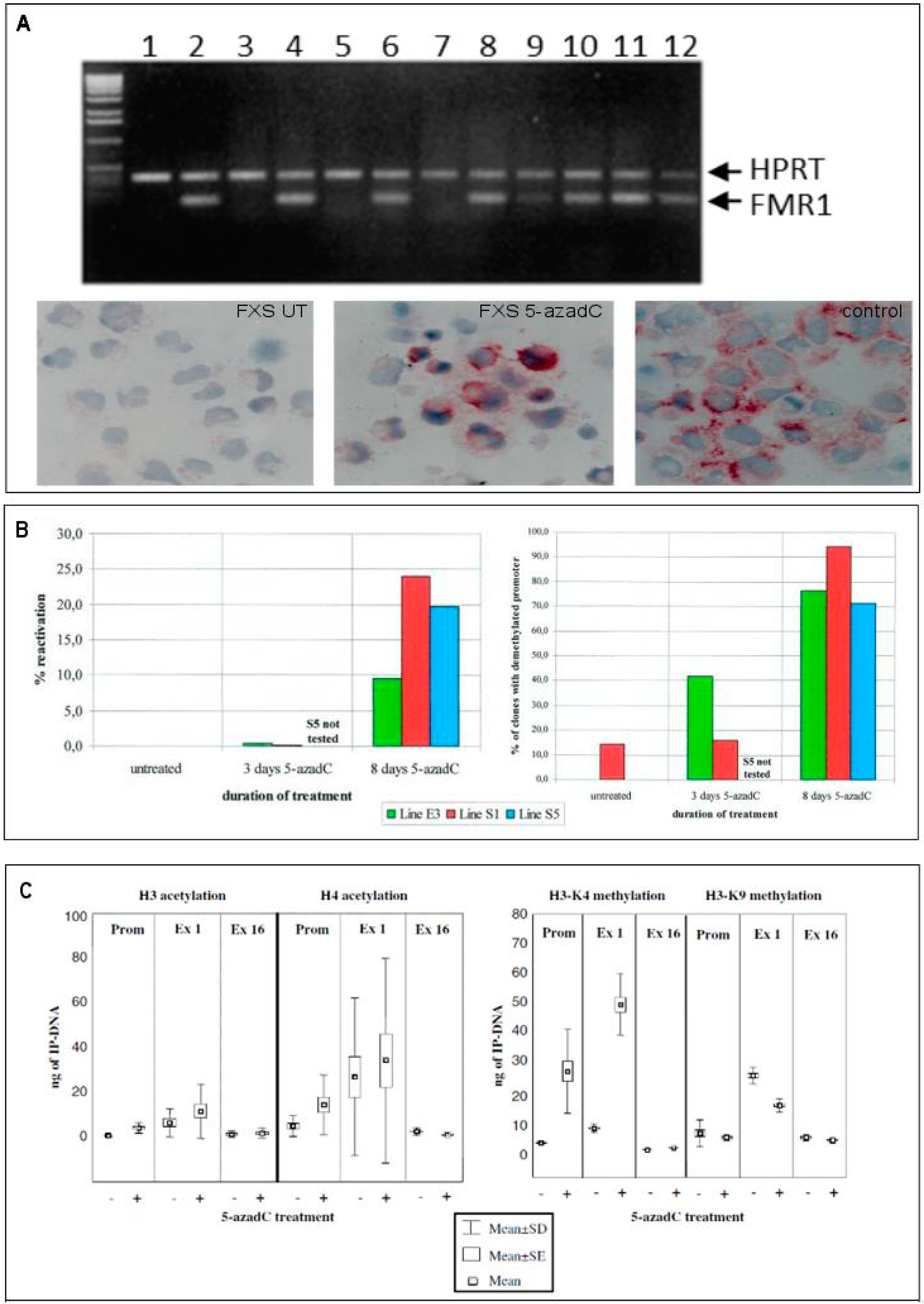
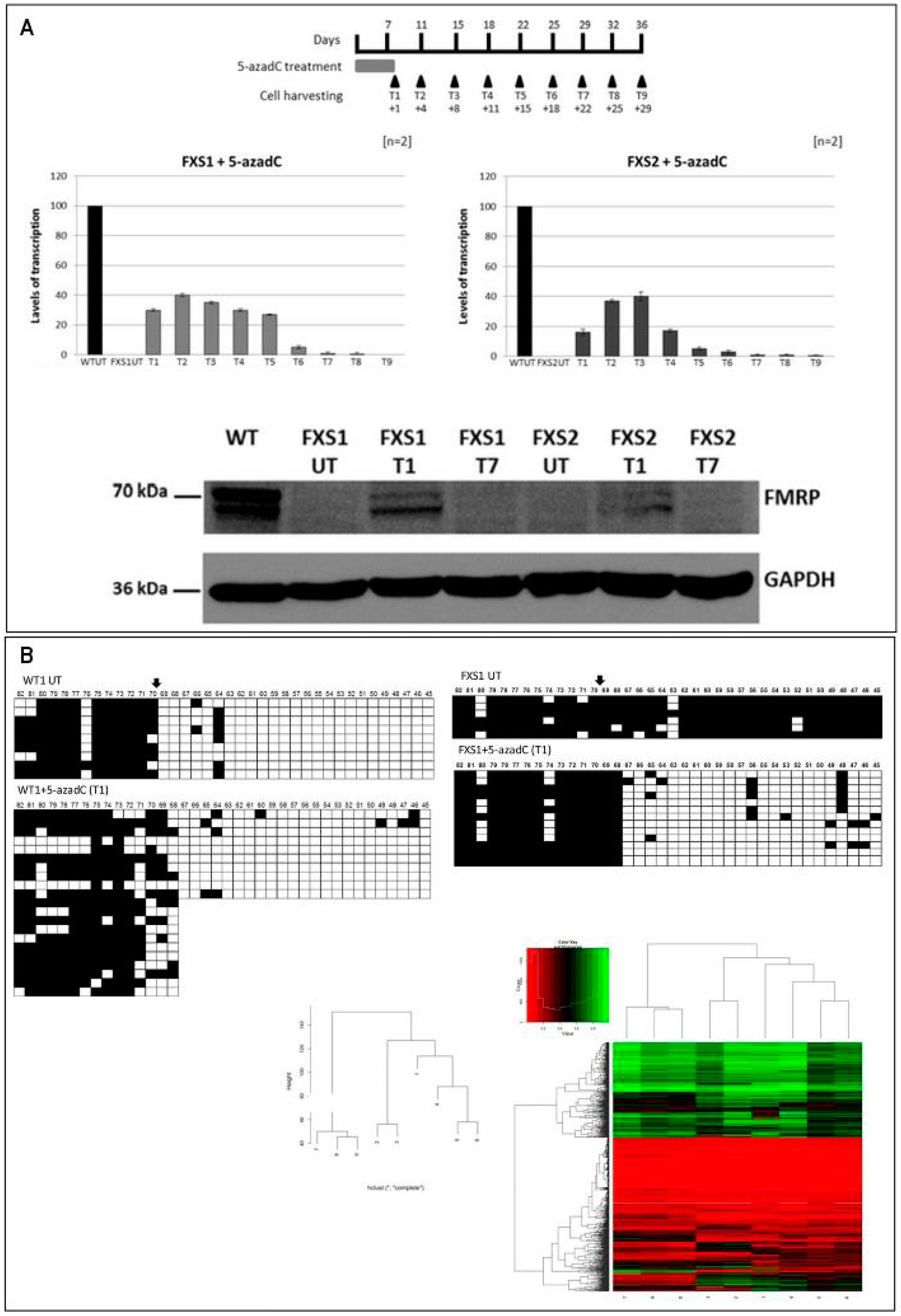
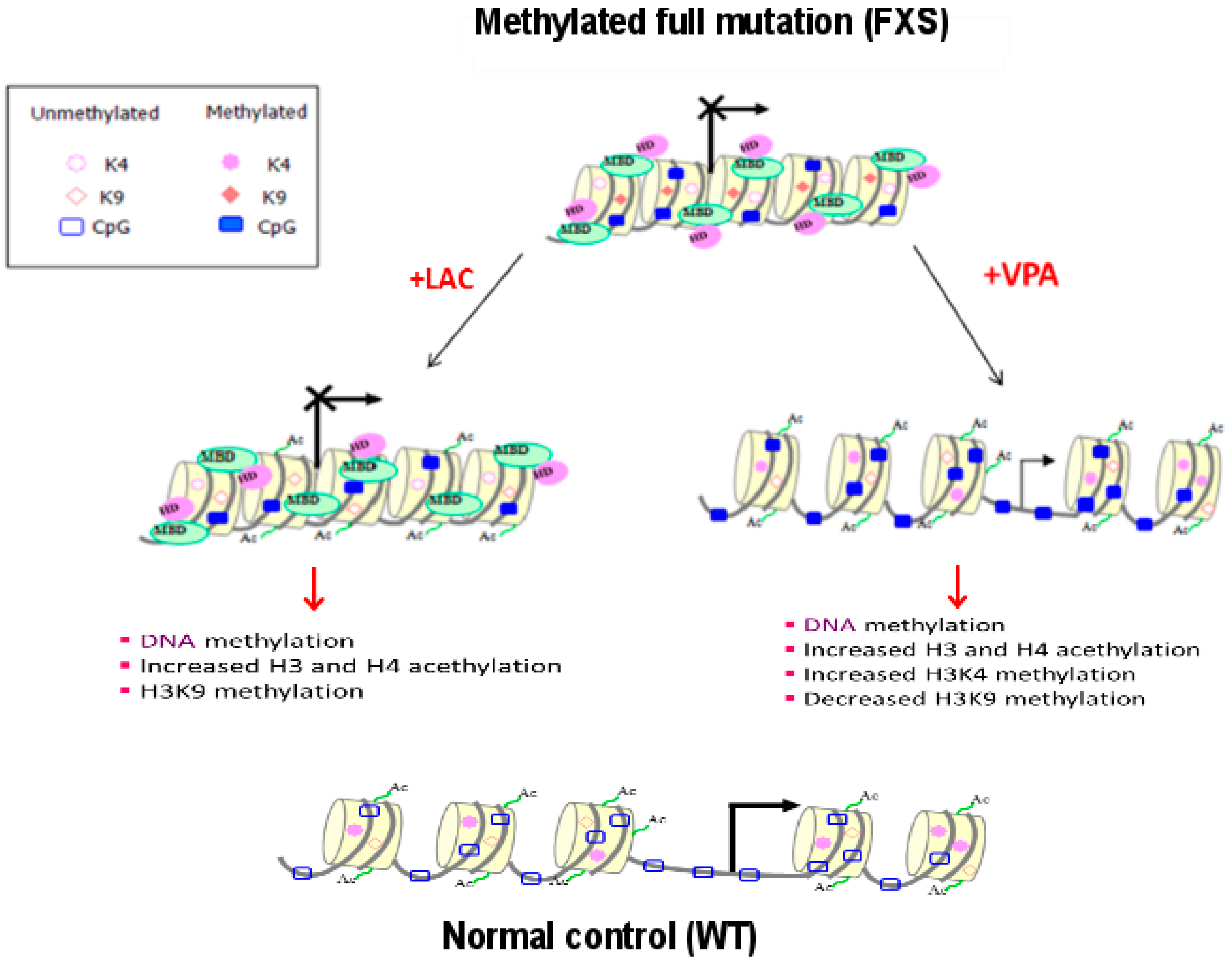
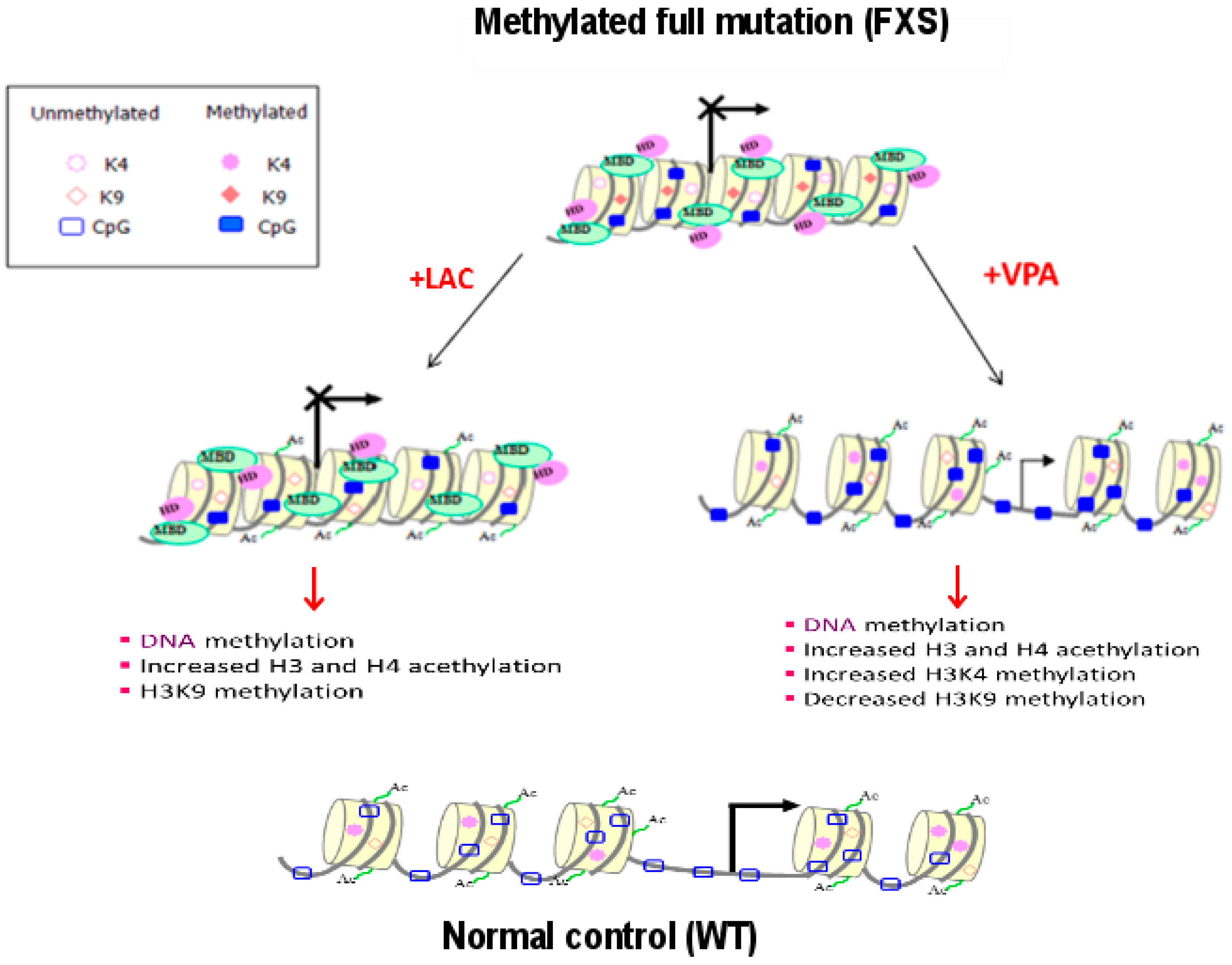
2.2. Epigenetic Treatments
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crawford, D.C.; Acuña, J.M.; Sherman, S.L. FMR1 and the Fragile X syndrome: Human genome epidemiology review. Genet. Med. 2001, 3, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Swain, R.A.; Christmon, C.A.; Chakravarti, A.; Weiler, I.J.; Greenough, W.T. Evidence for altered Fragile-X mental retardation protein expression in response to behavioral stimulation. Neurobiol. Learn. Mem. 2000, 74, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of Fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. A 2014, 164, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, YH.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in Fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Fernández, E.; Rajan, N.; Bagni, C. The FMRP regulon: From targets to disease convergence. Front. Neurosci. 2013, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, F.; Tabolacci, E.; Neri, G. The FRAXopathies: Definition, overview, and update. Am. J. Med. Genet. A 2011, 155, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Hayward, B.E.; Kumari, D.; Lokanga, R.A.; Sciascia, N.; Zhao, X.N. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet. 2014, 5, 226. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J.; Hagerman, R.J. Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Smeets, H.J.; Smits, A.P.; Verheij, C.E.; Theelen, J.P.; Willemsen, R.; van de Burgt, I.; Hoogeveen, A.T.; Oosterwijk, J.C.; Oostra, B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995, 4, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, R.; Tabolacci, E.; Zalfa, F.; Zito, I.; Terracciano, A.; Moscato, U.; Bagni, C.; Oostra, B.; Chiurazzi, P.; Neri, G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Moscato, U.; Zalfa, F.; Bagni, C.; Chiurazzi, P.; Neri, G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008, 16, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in Fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; Eiges, R. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E.; Holden, J.J.A.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Brackett, D.M.; Qing, F.; Amieux, P.S.; Sellers, D.L.; Horner, P.J.; Morris, D.R. FMR1 transcript isoforms: Association with polyribosomes; regional and developmental expression in mouse brain. PLoS ONE 2013, 8, e58296. [Google Scholar] [CrossRef] [PubMed]
- Naumann, A.; Hochstein, N.; Weber, S.; Fanning, E.; Doefler, W. A distinct DNA-methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in Fragile X syndrome. Am. J. Hum. Genet. 2009, 85, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Lanni, S.; Goracci, M.; Borrelli, L.; Mancano, G.; Chiurazzi, P.; Moscato, U.; Ferré, F.; Helmer-Citterich, M.; Tabolacci, E.; Neri, G. Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet. 2013, 9, e1003601. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Tassone, F.; Schwartz, P.H.; Sahota, P.; Hagerman, P.J. Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum. Mol. Genet. 2004, 13, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Lopez Posadas, B.; Pan, R.; Raske, C.; Hagerman, P.J.; Tassone, F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.L.; Raske, C.; Tassone, F.; Garcia-Arocena, D.; Hershey, J.W.; Hagerman, P.J. Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res. 2009, 37, 6896–6904. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with Fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.L.; Jacobs, P.A.; Morton, N.E.; Froster-Iskenius, U.; Howard-Peebles, P.N.; Nielsen, K.B.; Partington, M.W.; Sutherland, G.R.; Turner, G.; Watson, M. Further segregation analysis of the Fragile X syndrome with special reference to transmitting males. Hum. Genet. 1985, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Gutekunst, C.A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [PubMed]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, A.; Siomi, M.C.; Siomi, H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002, 16, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.; Reenan, R.A.; Jongens, T.A. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Ceman, S.; O’Donnell, W.T.; Reed, M.; Patton, S.; Pohl, J.; Warren, S.T. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum. Mol. Genet. 2003, 12, 3295–3305. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O'Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; Darnell, R.B.; Warren, S.T. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in Fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
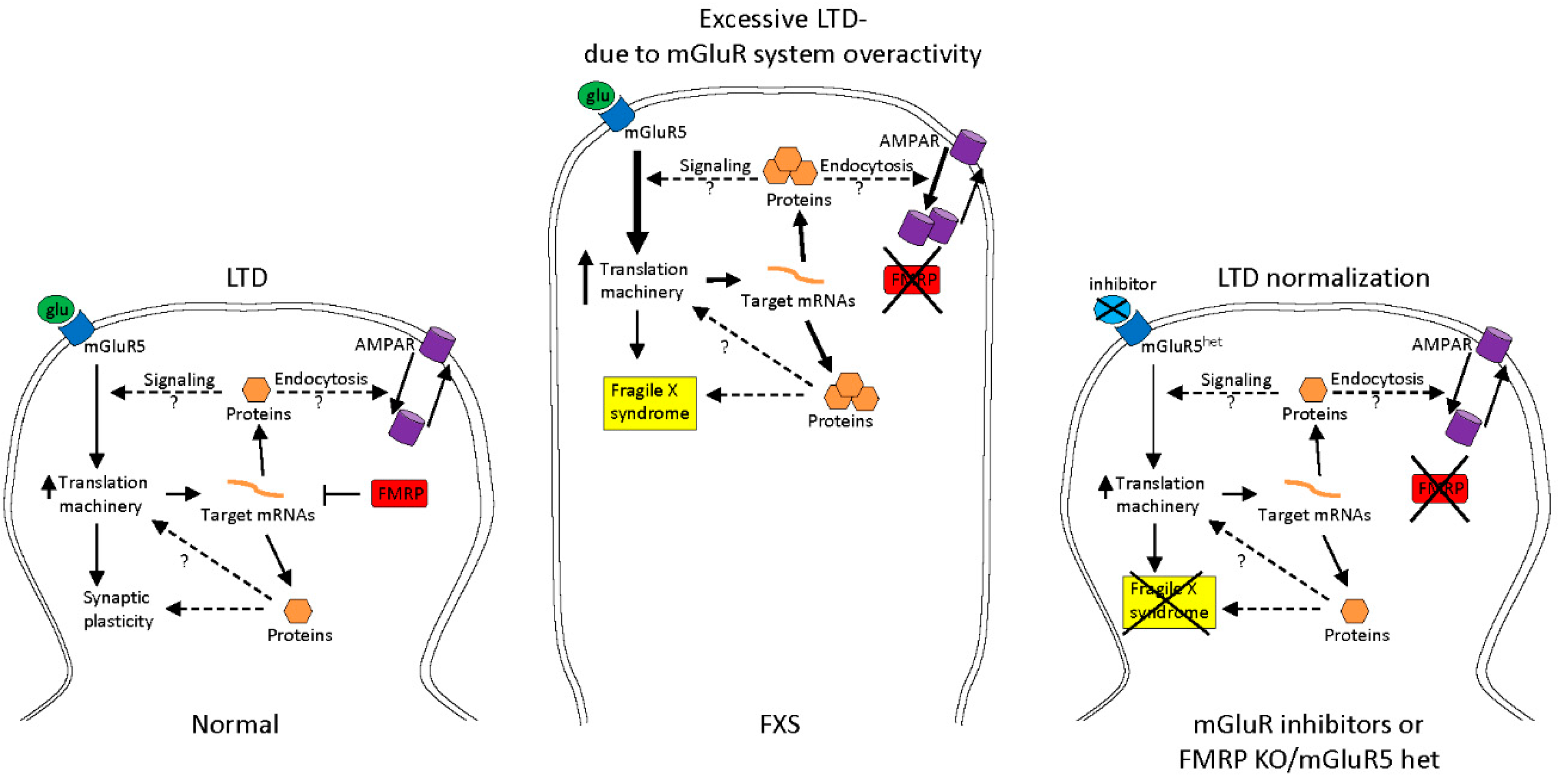
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, Y.; Ye, L.; Liu, B.; Liu, K.; Chen, J.; Xue, Q. Establishment of a hepatocellular carcinoma cell line with unique metastatic characteristics through in vivo selection and screening for metastasis-related genes through cDNA microarray. J. Cancer Res. Clin. Oncol. 2003, 129, 43–51. [Google Scholar] [PubMed]
- Liu, Y.; Zhu, X.; Zhu, J.; Liao, S.; Tang, Q.; Liu, K.; Guan, X.; Zhang, J.; Feng, Z. Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray. Oncol. Rep. 2007, 18, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Pedersen, S.; Hasle, H.; Olsen, J.H.; Friedrich, U. Evidence of decreased risk of cancer in individuals with fragile X. Am. J. Med. Genet. 2001, 103, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Kalkunte, R.; Macarthur, D.; Morton, R. Glioblastoma in a boy with fragile X: An unusual case of neuroprotection. Arch. Dis. Child. 2007, 92, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Lucá, R.; Averna, M.; Zalfa, F.; Vecchi, M.; Bianchi, F.; La Fata, G.; Del Nonno, F.; Nardacci, R.; Bianchi, M.; Nuciforo, P.; et al. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol. Med. 2013, 5, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stützer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epigenetics, Fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. A 2013, 161, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [PubMed]
- Tabolacci, E.; Pietrobono, R.; Moscato, U.; Oostra, B.A.; Chiurazzi, P.; Neri, G. Differential epigenetic modifications in the FMR1 gene of the Fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005, 13, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in Fragile X syndrome. Hum. Mol. Genet. 2010, 19, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; Pandey, U.B.; Di Prospero, N.A.; Min, K.T.; Taylor, J.P.; Paulson, H.L. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010, 6, e1001240. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Taylor, A.K.; Bridge, J.A. FMR1 fully expanded mutation with minimal methylation in a high functioning fragile X male. J. Med. Genet. 1996, 33, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Handa, V.; Saha, T.; Usdin, K. The Fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003, 31, 6243–6248. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 2007, 13, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; Ben-Yosef, D. Developmental study of Fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of Fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Goracci, M.; Lanni, S.; Mancano, G.; Palumbo, F.; Chiurazzi, P.; Neri, G.; Tabolacci, E. Defining the role of the CGGBP1 protein in FMR1 gene expression. Eur. J. Hum. Genet. 2016, 24, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Faghihi, M.A.; Modarresi, F.; Brothers, S.P.; Wahlestedt, C. A novel RNA transcript with antiapoptotic function is silenced in Fragile X syndrome. PLoS ONE 2008, 3, e1486. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Peschansky, V.J.; Barbouth, D.; Mehta, A.; Silva, J.P.; Wahlestedt, C. Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum. Genet. 2014, 133, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, X.; Glass, C.K.; Rosenfeld, M.G. The long arm of long noncoding RNAs: Roles as sensors regulating gene transcriptional programs. Cold Spring Harb. Perspect. Biol. 2011, 3, a003756. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Sanz, L.A.; Chédin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Kumari, D. Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front. Genet. 2015, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and Fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Curie, A.; des Portes, V.; Torrioli, M.G.; Berry-Kravis, E.; Hagerman, R.J.; Ramos, F.J.; Cornish, K.; He, Y.; Paulding, C.; et al. Epigenetic modification of the FMR1 gene in Fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 2011, 3, 64ra1. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Pirozzi, F.; Gomez-Mancilla, B.; Gasparini, F.; Neri, G. The mGluR5 antagonist AFQ056 does not affect methylation and transcription of the mutant FMR1 gene in vitro. BMC Med. Genet. 2012, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Oostra, B.A. Fragile X syndrome: From protein function to therapy. Am. J. Med. Genet. A 2013, 161, 2809–2821. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.L.; Davenport, M.H.; Erickson, C.A. Emerging pharmacologic treatment options for Fragile X syndrome. Appl. Clin. Genet. 2015, 8, 75–93. [Google Scholar] [PubMed]
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Willemsen, R.; Oostra, B.A.; Neri, G. In vitro reactivation of the FMR1 gene involved in Fragile X syndrome. Hum. Mol. Genet. 1998, 7, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, R.; Pomponi, M.G.; Tabolacci, E.; Oostra, B.; Chiurrazi, P.; Neri, G. Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 2002, 30, 3278–3285. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferrè, F.; Helmer-Citterich, M.; Neri, G. Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in Fragile X syndrome cells. Epigenet. Chromatin 2016, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Dickson, J.; Din, S.; Macleod, K.; Jodrell, D.; Ramsahoye, B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010, 38, 4313–4324. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Pietrobono, R.; Bakker, C.E.; Neri, G.; Oostra, B.A. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999, 8, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; De Pascalis, I.; Accadia, M.; Terraciano, A.; Moscato, U.; Chiurazzi, P.; Neri, G. Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet. Genom. 2008, 18, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.H.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of Valproic Acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Torrioli, M.; Vernacotola, S.; Setini, C.; Bevilacqua, F.; Martinelli, D.; Snape, M.; Hutchison, J.A.; Di Raimo, F.R.; Tabolacci, E.; Neri, G. Treatment with valproic acid ameliorates ADHD symptoms in Fragile X syndrome boys. Am. J. Med. Genet. A 2010, 152, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Torrioli, M.G.; Vernacotola, S.; Peruzzi, L.; Tabolacci, E.; Mila, M.; Militerni, R.; Musumeci, S.; Ramos, F.J.; Frontera, M.; Sorge, G.; et al. A double-blind, parallel, multicenter comparison of L-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in Fragile X syndrome boys. Am. J. Med. Genet. A 2008, 146, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in Fragile X iPSC-derived neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | FXS | UFM | |
|---|---|---|---|
| DNA methylation | − (absent) | + (present) | − (absent) |
| H3 and H4 acetylation | + | − | + |
| H3K4 methylation | + | − | + |
| H3K9 methylation | − | + | +/− |
| H3K27 dimethylation | + | − | + |
| H3K27 trimethylation | − | + | − |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, E.; Palumbo, F.; Nobile, V.; Neri, G. Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome. Genes 2016, 7, 49. https://doi.org/10.3390/genes7080049
Tabolacci E, Palumbo F, Nobile V, Neri G. Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome. Genes. 2016; 7(8):49. https://doi.org/10.3390/genes7080049
Chicago/Turabian StyleTabolacci, Elisabetta, Federica Palumbo, Veronica Nobile, and Giovanni Neri. 2016. "Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome" Genes 7, no. 8: 49. https://doi.org/10.3390/genes7080049
APA StyleTabolacci, E., Palumbo, F., Nobile, V., & Neri, G. (2016). Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome. Genes, 7(8), 49. https://doi.org/10.3390/genes7080049