
microRNA Expression in Sentinel Nodes from Progressing Melanoma Patients Identifies Networks Associated with Dysfunctional Immune Response

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Microarray Profiling and Data Analysis
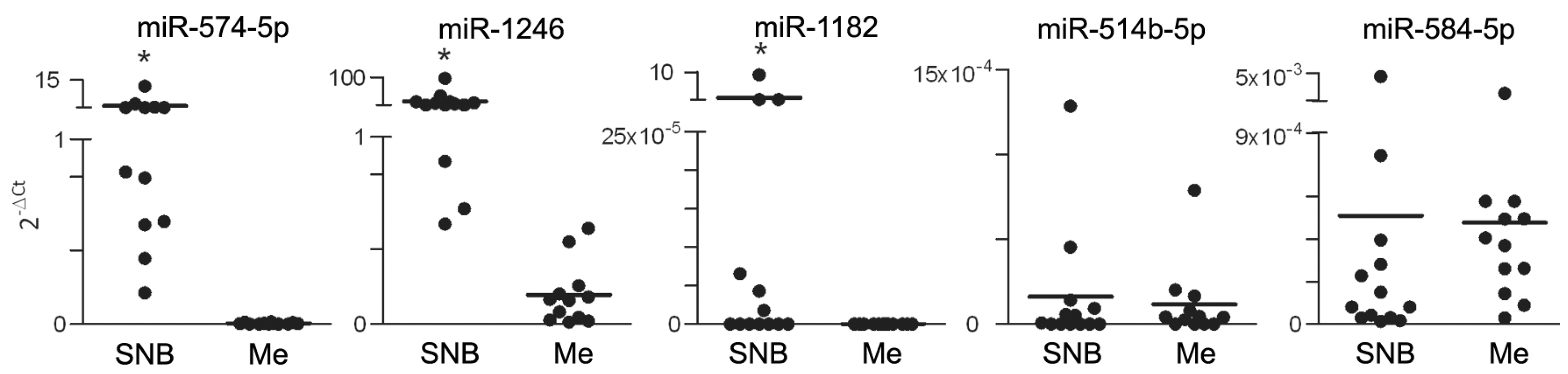
2.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR) Analysis
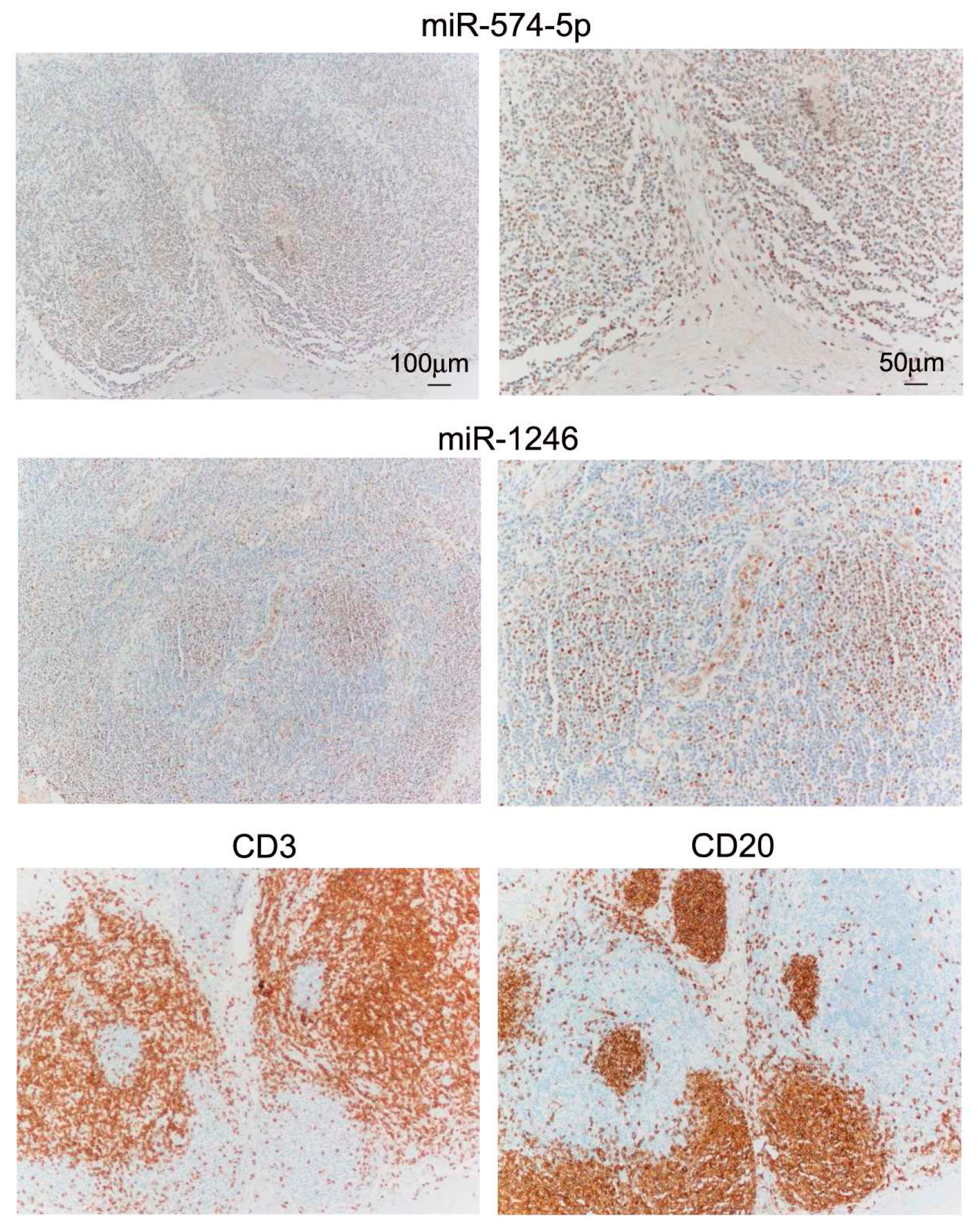
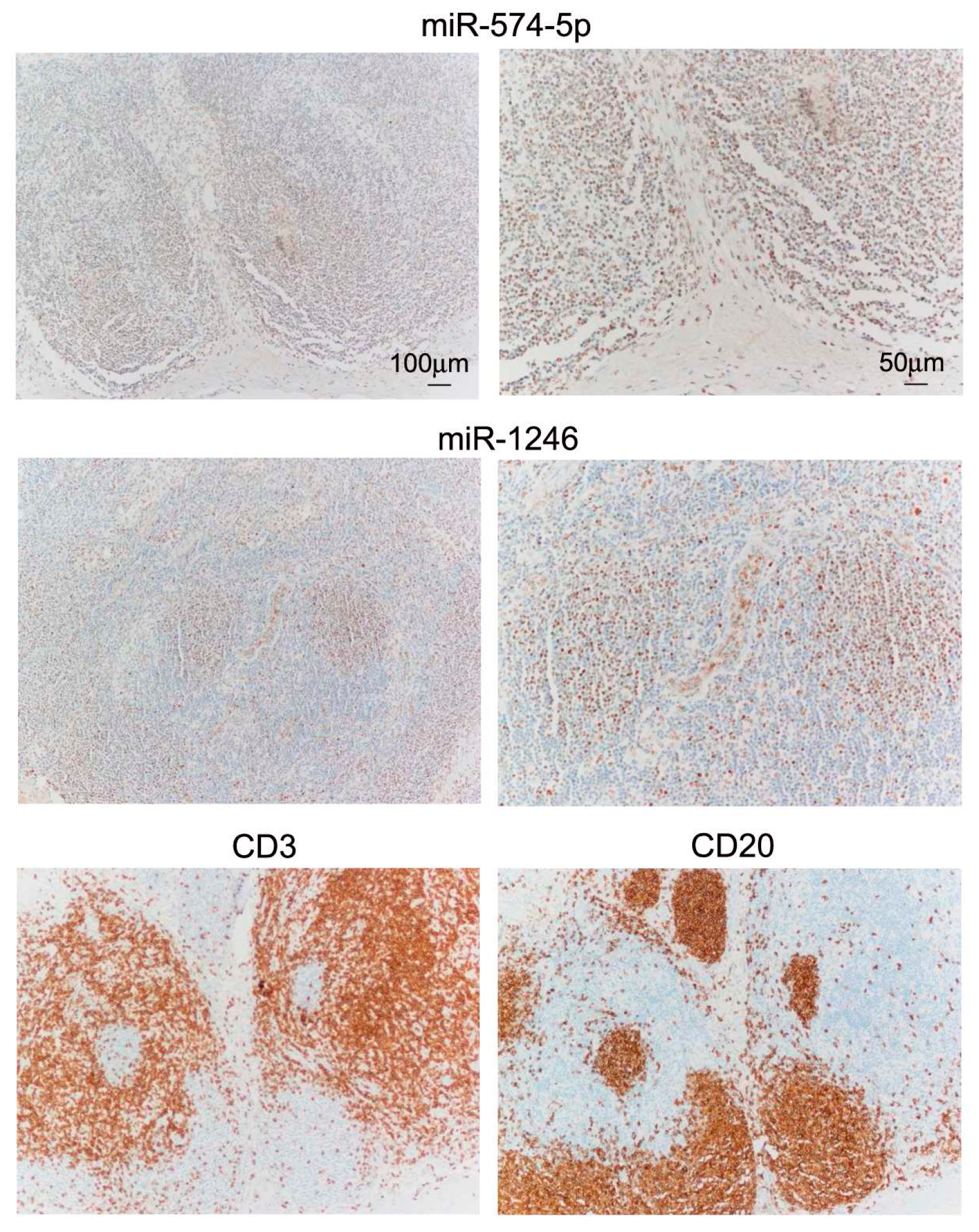
2.4. miR In Situ Hybridization (ISH) and Tissue Immunostaining
2.5. In vitro Lymphocyte Cultures and Fluorescence Activated Cell Sorting (FACS) Analysis
3. Results
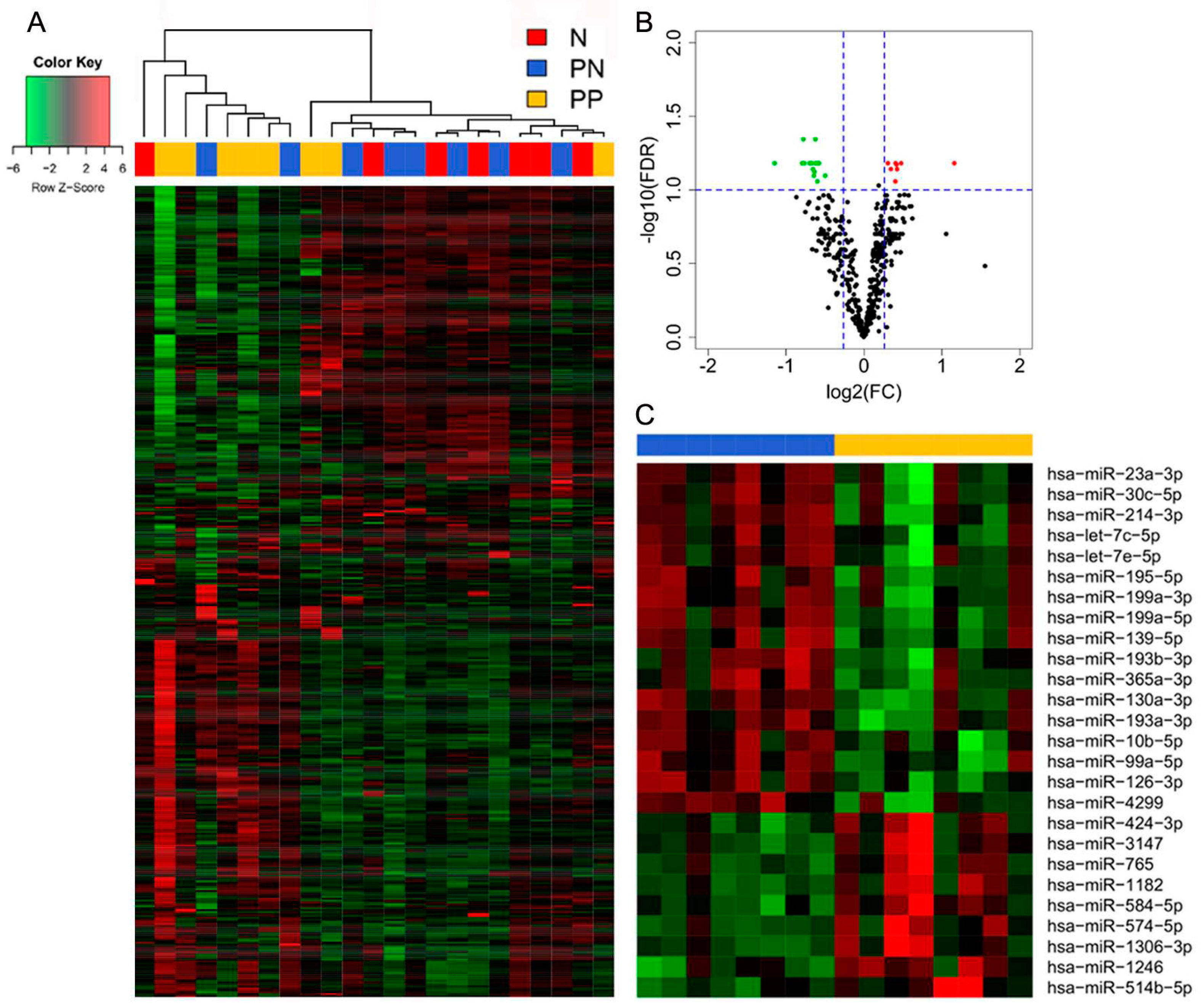
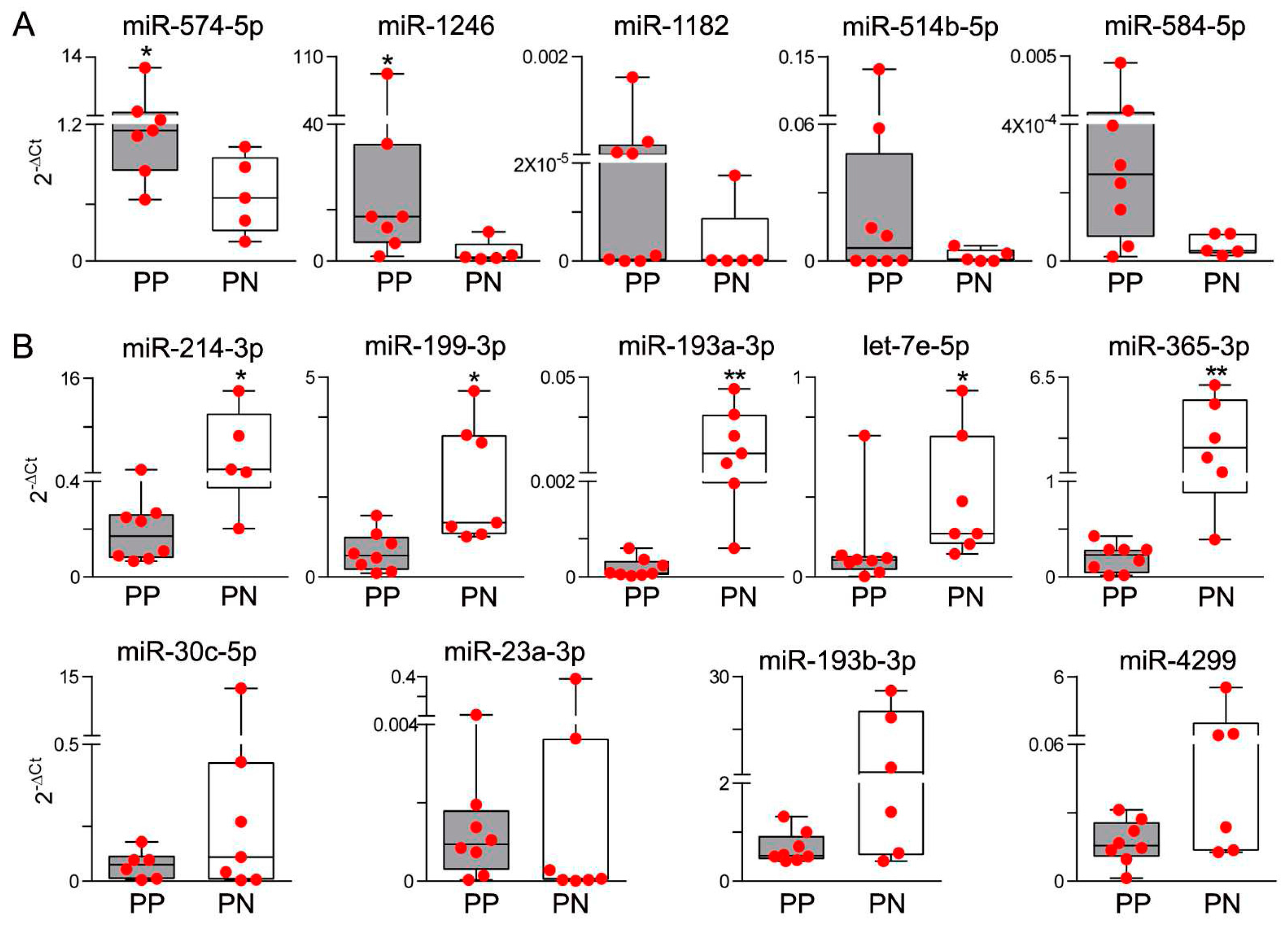
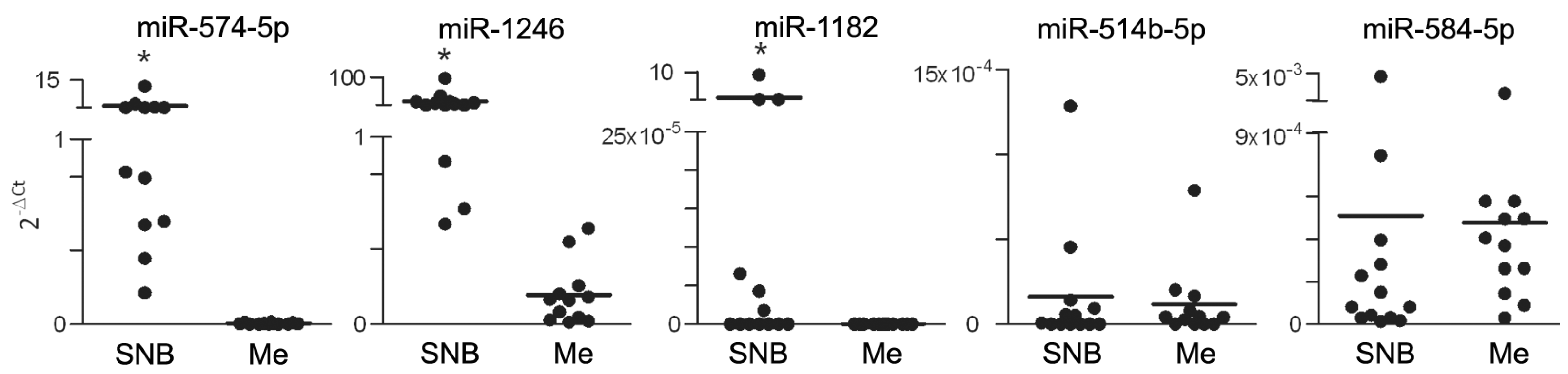
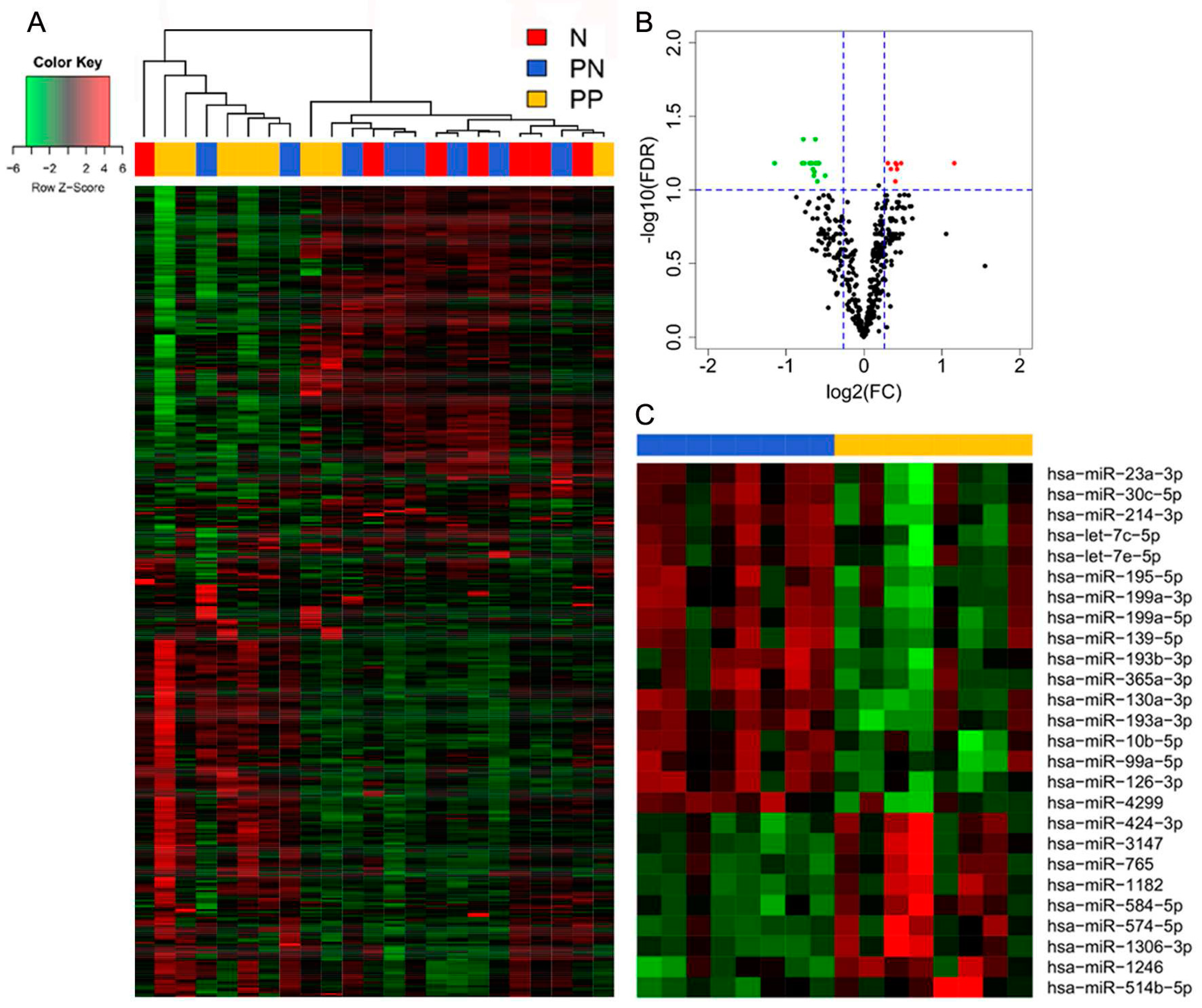
3.1. Differential miR Expression Characterizes SNB in Patients with Progressive Disease
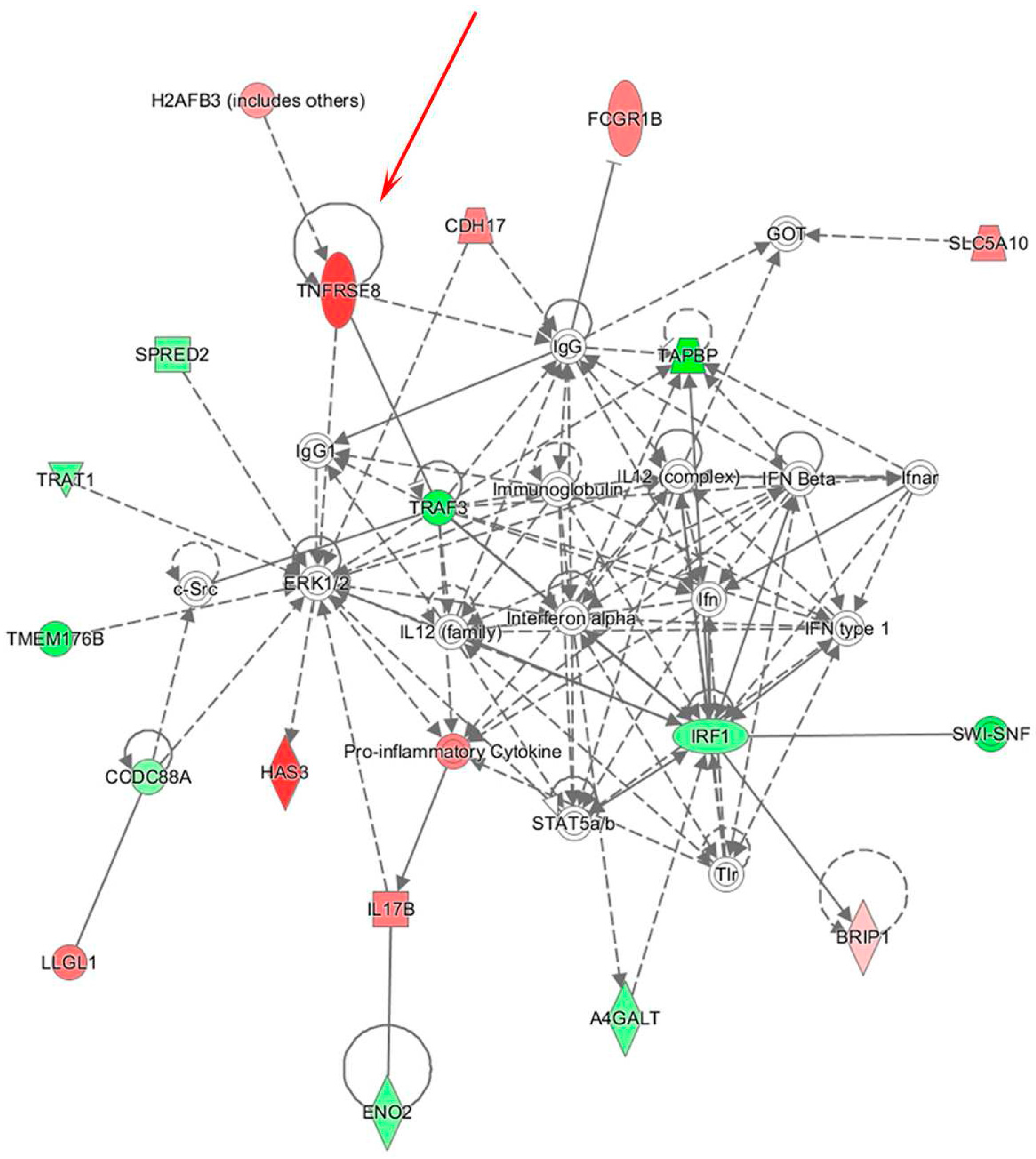
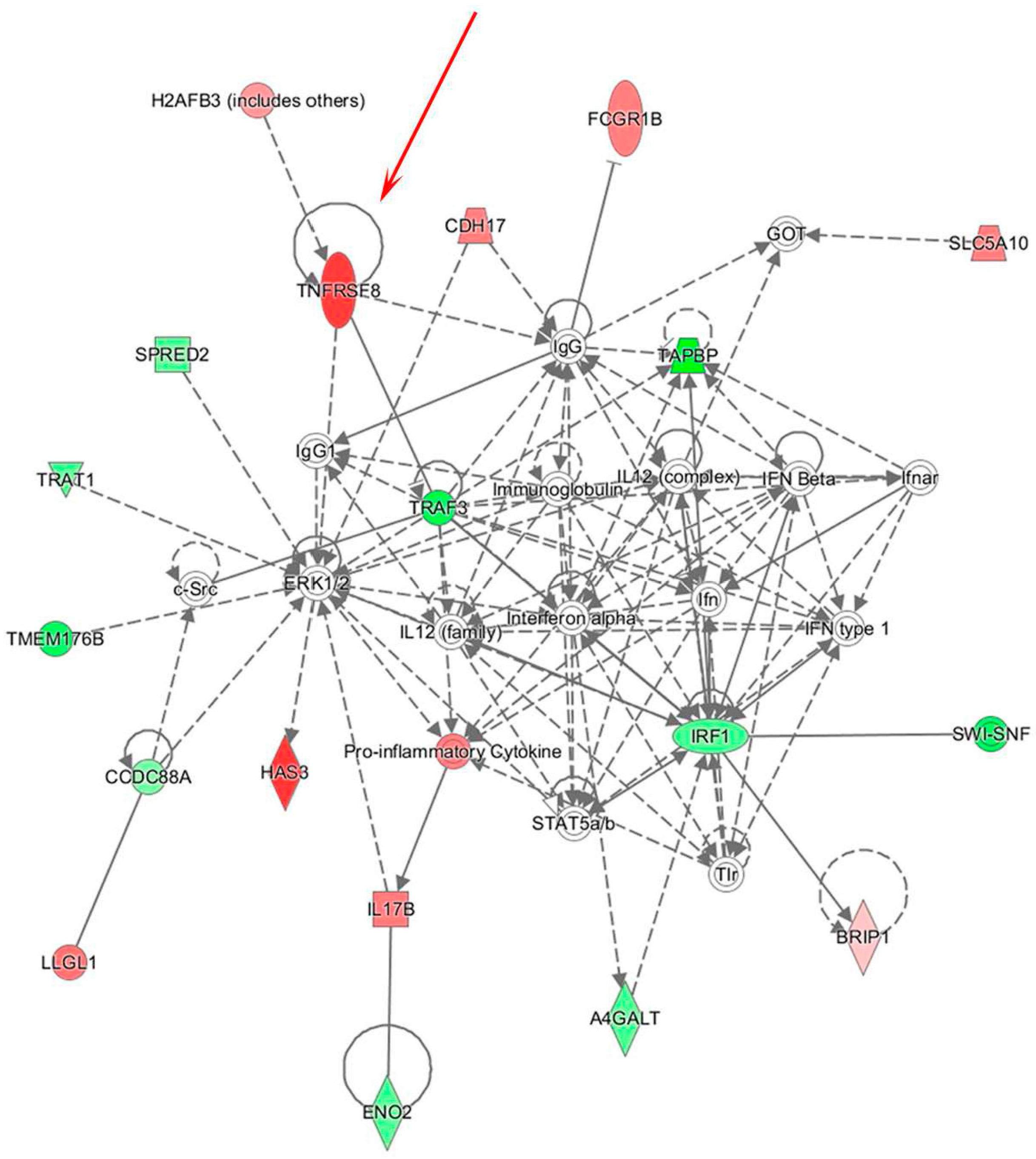
3.2. Integrative Analysis of miR and Gene Expression Data Identifies Regulatory Networks in the Lymph Node
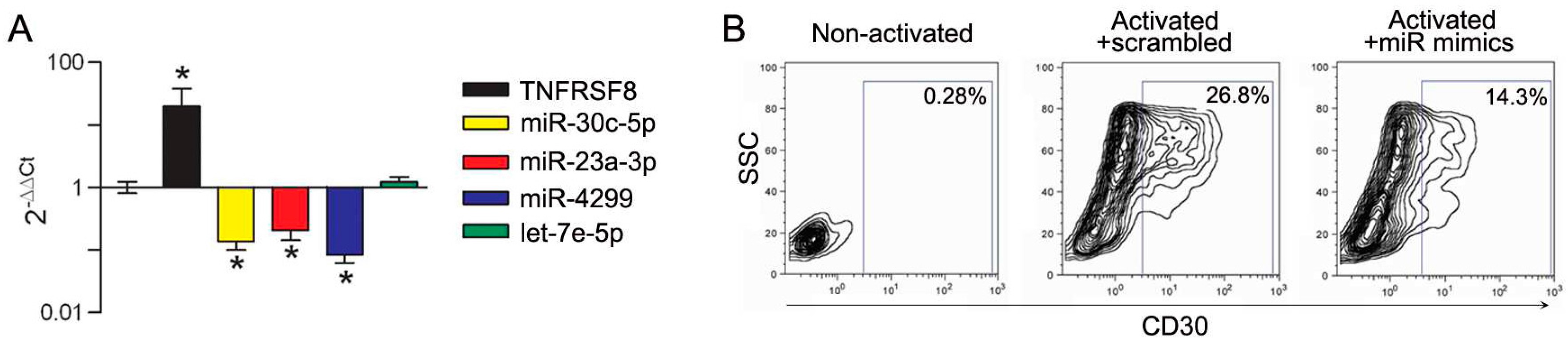
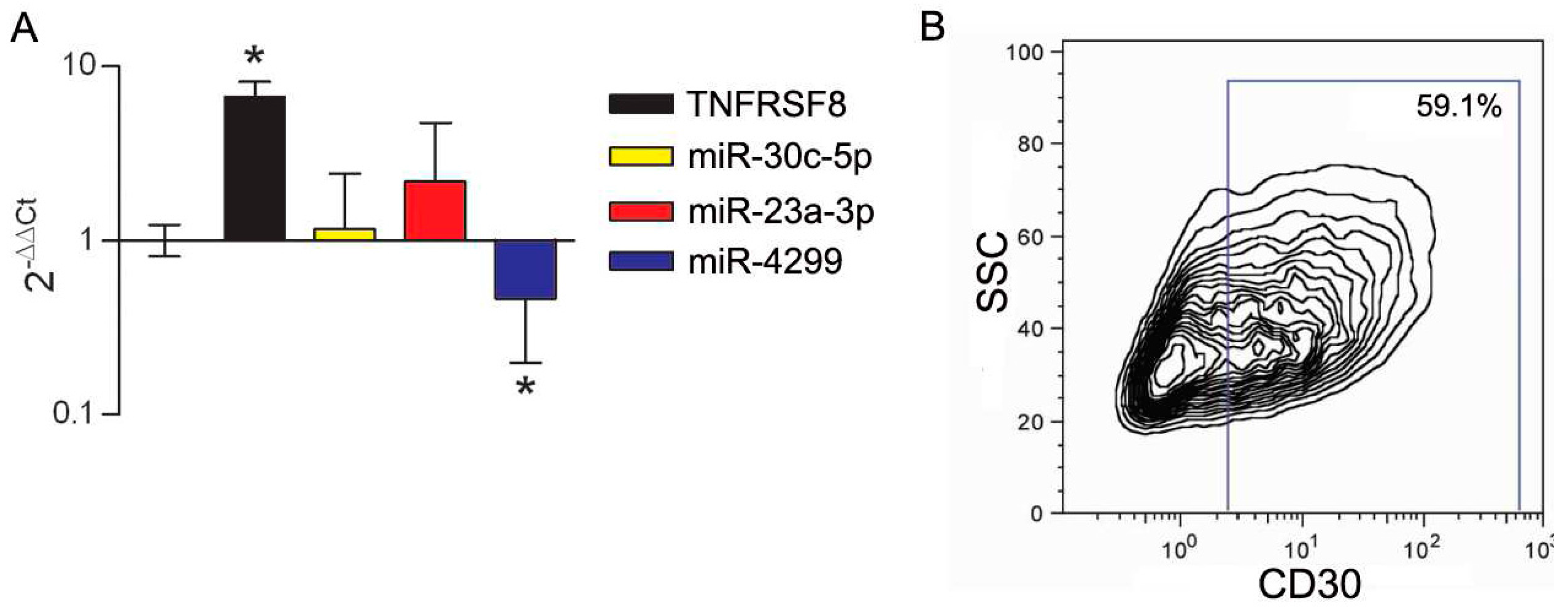
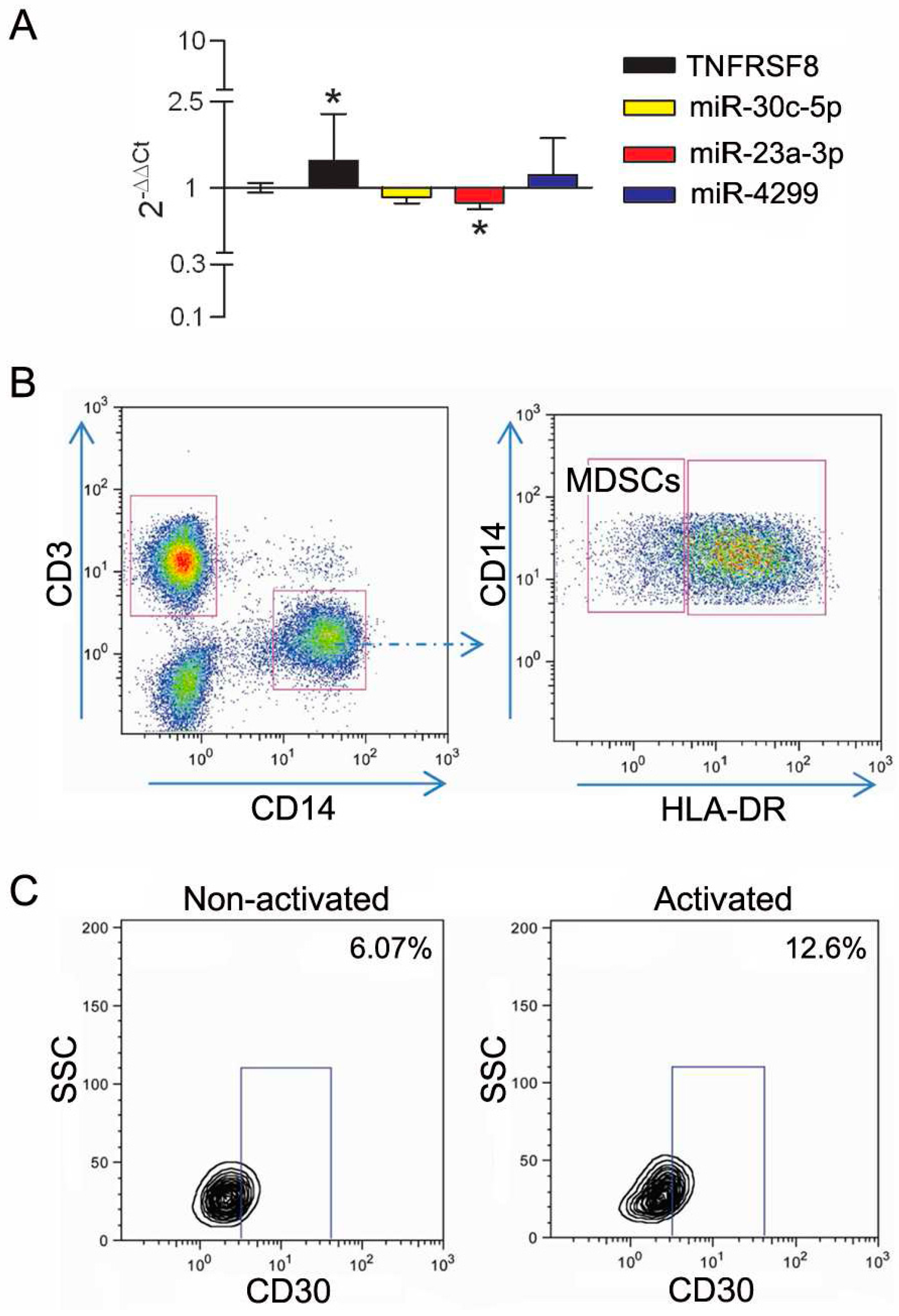
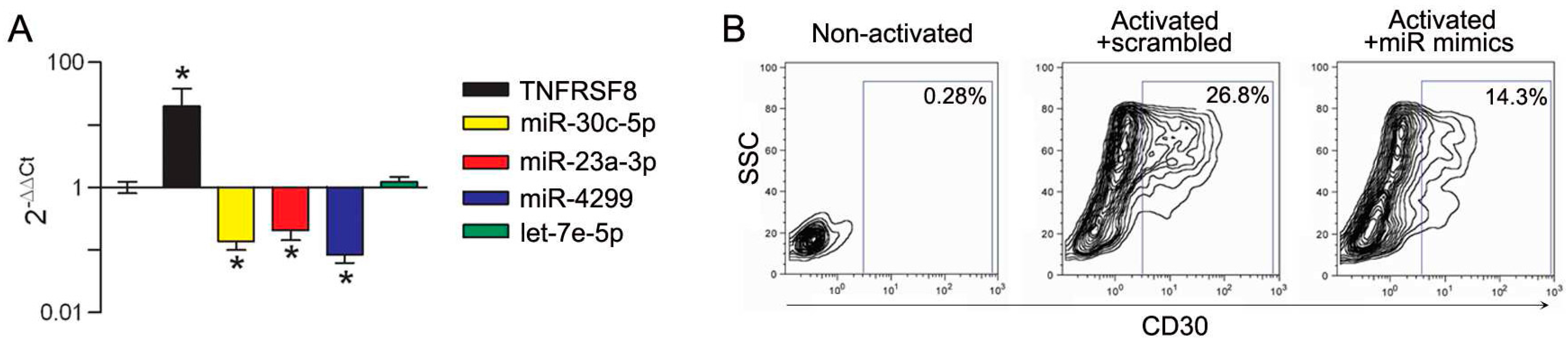
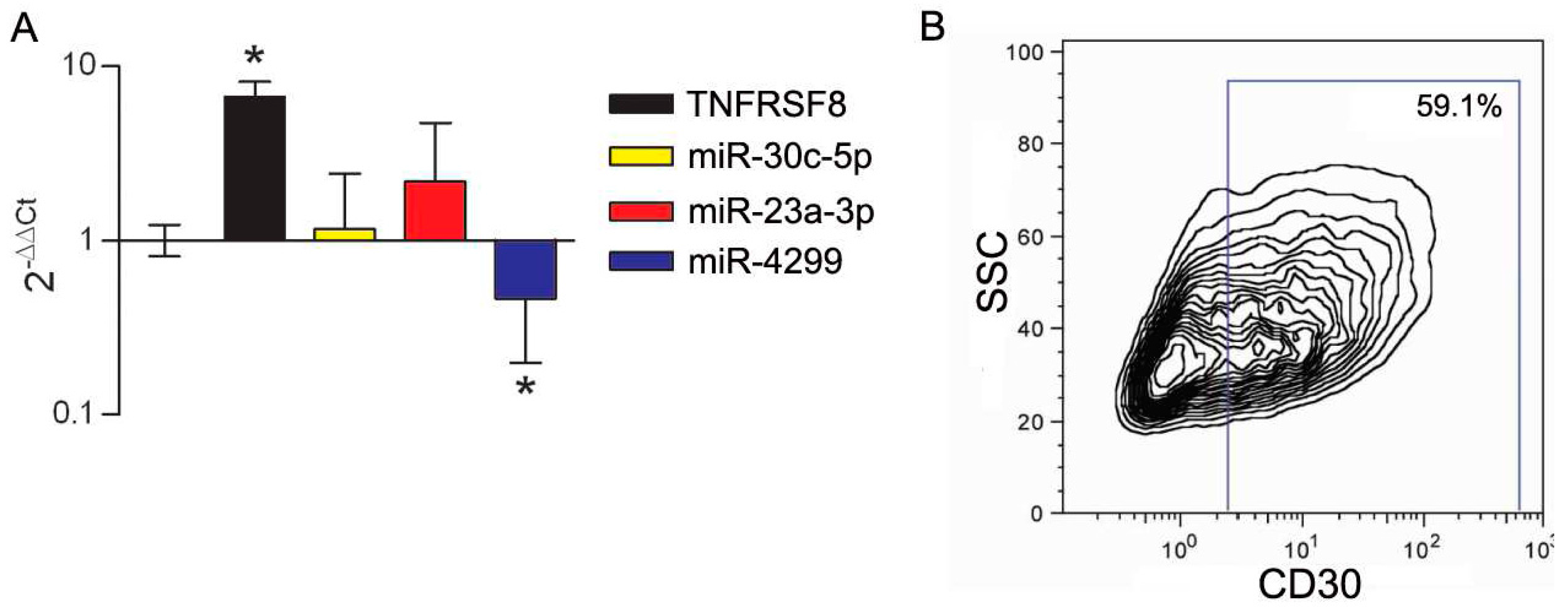
3.3. Modulation of miRs during In Vitro Induction of CD30 in Immune Cells
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartlett, E.K.; Karakousis, G.C. Current Staging and Prognostic Factors in Melanoma. Surg. Oncol. Clin. N. Am. 2015, 24, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New Regulators of Immune Cell Development and Function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef] [PubMed]
- O′Connell, R.M.; Zhao, J.L.; Rao, D.S. MicroRNA Function in Myeloid Biology. Blood 2011, 118, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Jeker, L.T.; Bluestone, J.A. MicroRNA Regulation of T-Cell Differentiation and Function. Immunol. Rev. 2013, 253, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, P.; Wang, X.F. Microenvironmental Regulation of Cancer Metastasis by miRNAs. Trends Cell Biol. 2014, 24, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Katsura, A.; Matsuyama, H.; Miyazono, K. MicroRNA Regulons in Tumor Microenvironment. Oncogene 2015, 34, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Vallacchi, V.; Vergani, E.; Camisaschi, C.; Deho, P.; Cabras, A.D.; Sensi, M.; De Cecco, L.; Bassani, N.; Ambrogi, F.; Carbone, A.; et al. Transcriptional Profiling of Melanoma Sentinel Nodes Identify Patients with Poor Outcome and Reveal an Association of CD30(+) T Lymphocytes with Progression. Cancer Res. 2014, 74, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.R.; Su, A.; Itakura, E.; Wen, D.R. Is Sentinel Node Susceptibility to Metastases Related to Nodal Immune Modulation? Cancer J. 2015, 21, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, M.; Castelli, C.; Rivoltini, L. Immune Response Markers in Sentinel Nodes may Predict Melanoma Progression. Oncoimmunology 2014, 3, e28498. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Gualeni, A.V.; Volpi, C.C.; Carbone, A.; Gloghini, A. A Novel Semi-Automated in Situ Hybridisation Protocol for microRNA Detection in Paraffin Embedded Tissue Sections. J. Clin. Pathol. 2015, 68, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4(+)CD25(High)Foxp3(+) Regulatory T Cells that are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M. MicroRNAs as Potential Regulators of Myeloid-Derived Suppressor Cell Expansion. Innate Immun. 2014, 20, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Smigielska-Czepiel, K.; van den Berg, A.; Jellema, P.; van der Lei, R.J.; Bijzet, J.; Kluiver, J.; Boots, A.M.; Brouwer, E.; Kroesen, B.J. Comprehensive Analysis of miRNA Expression in T-Cell Subsets of Rheumatoid Arthritis Patients Reveals Defined Signatures of Naive and Memory Tregs. Genes Immun. 2014, 15, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Ballarino, M.; Sorrentino, A.; Sthandier, O.; De Angelis, F.G.; Marchioni, M.; Masella, B.; Guarini, A.; Fatica, A.; Peschle, C.; et al. The Interplay between the Master Transcription Factor PU.1 and miR-424 Regulates Human monocyte/macrophage Differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 19849–19854. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, S.; MacPherson, C.R.; Essack, M.; Kaur, M.; Schaefer, U.; Suzuki, H.; Hayashizaki, Y.; Bajic, V.B. Deciphering the Transcriptional Circuitry of microRNA Genes Expressed during Human Monocytic Differentiation. BMC Genom. 2009. [Google Scholar] [CrossRef] [PubMed]
- Pedranzini, L.; Mottadelli, F.; Ronzoni, S.; Rossella, F.; Ferracin, M.; Magnani, I.; Roversi, G.; Colapietro, P.; Negrini, M.; Pelicci, P.G.; et al. Differential Cytogenomics and miRNA Signature of the Acute Myeloid Leukaemia Kasumi-1 Cell Line CD34+38− Compartment. Leuk. Res. 2010, 34, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Jindra, P.T.; Bagley, J.; Godwin, J.G.; Iacomini, J. Costimulation-Dependent Expression of microRNA-214 Increases the Ability of T Cells to Proliferate by Targeting Pten. J. Immunol. 2010, 185, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ma, H.; Hu, H.; Gao, L.; Wang, X.; Ma, J.; Gao, Q.; Liu, B.; Zhou, G.; Liang, C. Special Role of Foxp3 for the Specifically Altered microRNAs in Regulatory T Cells of HCC Patients. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Alemdehy, M.F.; Haanstra, J.R.; de Looper, H.W.; van Strien, P.M.; Verhagen-Oldenampsen, J.; Caljouw, Y.; Sanders, M.A.; Hoogenboezem, R.; de Ru, A.H.; Janssen, G.M.; et al. ICL-Induced miR139–3p and miR199a-3p have Opposite Roles in Hematopoietic Cell Expansion and Leukemic Transformation. Blood 2015, 125, 3937–3948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. Dicer Controls CD8+ T-Cell Activation, Migration, and Survival. Proc. Natl. Acad. Sci. USA 2010, 107, 21629–21634. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, C.S.; Kalra, V.K. Hypoxia-Mediated Expression of 5-Lipoxygenase-Activating Protein Involves HIF-1alpha and NF-kappaB and microRNAs 135a and 199a-5p. J. Immunol. 2010, 184, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Parlato, S.; Bruni, R.; Fragapane, P.; Salerno, D.; Marcantonio, C.; Borghi, P.; Tataseo, P.; Ciccaglione, A.R.; Presutti, C.; Romagnoli, G.; et al. IFN-Alpha Regulates Blimp-1 Expression Via miR-23a and miR-125b in both Monocytes-Derived DC and pDC. PLoS ONE 2013, 8, e72833. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA Let-7c Regulates Macrophage Polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.; Liu, W.; Liu, Y.; Xu, J. Polycomb-Mediated Loss of microRNA Let-7c Determines Inflammatory Macrophage Polarization Via PAK1-Dependent NF-kappaB Pathway. Cell Death Differ. 2015, 22, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chen, L.; Chen, G.; Hu, C.; Jiang, S.; Sevilla, J.; Wan, Y.; Sampson, J.H.; Zhu, B.; Li, Q.J. Targeting miR-23a in CD8+ Cytotoxic T Lymphocytes Prevents Tumor-Dependent Immunosuppression. J. Clin. Investig. 2014, 124, 5352–5367. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, M.; Haga, C.L.; Das, S.; Leu, C.M.; Hodson, D.; Josson, S.; Turner, M.; Cooper, M.D. MicroRNA 125b Inhibition of B Cell Differentiation in Germinal Centers. Int. Immunol. 2010, 22, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, J.; Bonin, M.; Walter, M.; Fend, F.; Bonzheim, I.; Quintanilla-Martinez, L. Next-Generation Sequencing Identifies Deregulation of microRNAs Involved in both Innate and Adaptive Immune Response in ALK+ ALCL. PLoS ONE 2015, 10, e0117780. [Google Scholar] [CrossRef] [PubMed]
- Grigoryev, Y.A.; Kurian, S.M.; Hart, T.; Nakorchevsky, A.A.; Chen, C.; Campbell, D.; Head, S.R.; Yates, J.R., 3rd; Salomon, D.R. MicroRNA Regulation of Molecular Networks Mapped by Global microRNA, mRNA, and Protein Expression in Activated T Lymphocytes. J. Immunol. 2011, 187, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Mets, E.; Van der Meulen, J.; Van Peer, G.; Boice, M.; Mestdagh, P.; Van de Walle, I.; Lammens, T.; Goossens, S.; De Moerloose, B.; Benoit, Y.; et al. MicroRNA-193b-3p Acts as a Tumor Suppressor by Targeting the MYB Oncogene in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2015, 29, 798–806. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and Pathological Roles for microRNAs in the Immune System. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Bisognin, A.; Saccoman, C.; Mannarelli, C.; Coppe, A.; Vannucchi, A.M.; Bortoluzzi, S. Small RNA Sequencing Uncovers New miRNAs and moRNAs Differentially Expressed in Normal and Primary Myelofibrosis CD34+ Cells. PLoS ONE 2015, 10, e0140445. [Google Scholar] [CrossRef] [PubMed]
- Trifari, S.; Pipkin, M.E.; Bandukwala, H.S.; Aijo, T.; Bassein, J.; Chen, R.; Martinez, G.J.; Rao, A. MicroRNA-Directed Program of Cytotoxic CD8+ T-Cell Differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 18608–18613. [Google Scholar] [CrossRef] [PubMed]
- Muta, H.; Podack, E.R. CD30: From Basic Research to Cancer Therapy. Immunol. Res. 2013, 57, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a New Subset of Myeloid Suppressor Cells in Peripheral Blood of Melanoma Patients with Modulation by a Granulocyte-Macrophage Colony-Stimulation Factor-Based Antitumor Vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.C., Jr.; Clemente, C.G.; Cascinelli, N. Tumor Infiltrating Lymphocytes in Lymph Node Melanoma Metastases: A Histopathologic Prognostic Indicator and an Expression of Local Immune Response. Lab. Investig. 1996, 74, 43–47. [Google Scholar] [PubMed]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-Infiltrating Lymphocyte Grade is an Independent Predictor of Sentinel Lymph Node Status and Survival in Patients with Cutaneous Melanoma. J. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef] [PubMed]
- Camisaschi, C.; Vallacchi, V.; Castelli, C.; Rivoltini, L.; Rodolfo, M. Immune Cells in the Melanoma Microenvironment Hold Information for Prediction of the Risk of Recurrence and Response to Treatment. Expert Rev. Mol. Diagn. 2014, 14, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-Genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer. Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.R.; Lee, J.; Itakura, E.; Leong, S.P.; Essner, R. Tumour-Induced Immune Modulation of Sentinel Lymph Nodes. Nat. Rev. Immunol. 2006, 6, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Messaoudene, M.; Perier, A.; Fregni, G.; Neves, E.; Zitvogel, L.; Cremer, I.; Chanal, J.; Sastre-Garau, X.; Deschamps, L.; Marinho, E.; et al. Characterization of the Microenvironment in Positive and Negative Sentinel Lymph Nodes from Melanoma Patients. PLoS ONE 2015, 10, e0133363. [Google Scholar] [CrossRef] [PubMed]
- Mohos, A.; Sebestyen, T.; Liszkay, G.; Plotar, V.; Horvath, S.; Gaudi, I.; Ladanyi, A. Immune Cell Profile of Sentinel Lymph Nodes in Patients with Malignant Melanoma—FOXP3+ Cell Density in Cases with Positive Sentinel Node Status is Associated with Unfavorable Clinical Outcome. J. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Jebbawi, F.; Fayyad-Kazan, H.; Merimi, M.; Lewalle, P.; Verougstraete, J.C.; Leo, O.; Romero, P.; Burny, A.; Badran, B.; Martiat, P.; et al. A microRNA Profile of Human CD8(+) Regulatory T Cells and Characterization of the Effects of microRNAs on Treg Cell-Associated Genes. J. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lui, W.O.; Fire, A.; Dadras, S.S. Profiling and Discovery of Novel miRNAs from Formalin-Fixed, Paraffin-Embedded Melanoma and Nodal Specimens. J. Mol. Diagn. 2009, 11, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.T.; Lo, C.M.; Wong, N.; Li, C.X.; Qi, X.; Liu, X.B.; Geng, W.; Yeung, O.W.; Ma, Y.Y.; Chan, S.C.; et al. Early-Phase Circulating miRNAs Predict Tumor Recurrence and Survival of Hepatocellular Carcinoma Patients After Liver Transplantation. Oncotarget 2016, 7, 19824–19839. [Google Scholar] [CrossRef] [PubMed]
- Armand-Labit, V.; Meyer, N.; Casanova, A.; Bonnabau, H.; Platzer, V.; Tournier, E.; Sansas, B.; Verdun, S.; Thouvenot, B.; Hilselberger, B.; et al. Identification of a Circulating MicroRNA Profile as a Biomarker of Metastatic Cutaneous Melanoma. Acta Derm. Venereol. 2016, 96, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kimbrough, C.W.; Egger, M.E.; McMasters, K.M.; Stromberg, A.J.; Martin, R.C.; Philips, P.; Scoggins, C.R. Molecular Staging of Sentinel Lymph Nodes Identifies Melanoma Patients at Increased Risk of Nodal Recurrence. J. Am. Coll. Surg. 2016, 222, 357–363. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Fold Change | FDR | p-Value |
|---|---|---|---|
| miR-574-5p | 2.237 | 0.066 | 0.001 |
| miR-1182 | 1.393 | 0.066 | 0.001 |
| miR-1246 | 1.393 | 0.066 | 0.002 |
| miR-514b-5p | 1.342 | 0.072 | 0.004 |
| miR-424-3p | 1.336 | 0.067 | 0.003 |
| miR-3147 | 1.326 | 0.066 | 0.002 |
| miR-765 | 1.324 | 0.088 | 0.005 |
| miR-1306-3p | 1.272 | 0.072 | 0.003 |
| miR-584-5p | 1.238 | 0.066 | 0.001 |
| miR-214-3p | −2.209 | 0.066 | 0.001 |
| miR-199a-3p a | −1.731 | 0.066 | 0.002 |
| miR-130a-3p | −1.711 | 0.045 | 0.000 |
| miR-195-5p | −1.699 | 0.066 | 0.002 |
| miR-30c-5p | −1.622 | 0.066 | 0.003 |
| miR-193a-3p | −1.592 | 0.066 | 0.002 |
| let-7c-5p | −1.572 | 0.072 | 0.003 |
| miR-23a-3p | −1.559 | 0.080 | 0.005 |
| miR-99a-5p | −1.552 | 0.075 | 0.004 |
| miR-199a-5p | −1.541 | 0.066 | 0.001 |
| miR-126-3p | −1.539 | 0.045 | 0.000 |
| miR-193b-3p | −1.519 | 0.066 | 0.003 |
| let-7e-5p | −1.512 | 0.088 | 0.005 |
| miR-365a-3p b | −1.503 | 0.066 | 0.001 |
| miR-10b-5p | −1.500 | 0.066 | 0.002 |
| miR-4299 | −1.490 | 0.066 | 0.002 |
| miR-139-5p | −1.410 | 0.080 | 0.004 |
| Gene Name | Regulation | Immune Compartment | Functions in Immune System | Reference |
|---|---|---|---|---|
| miR-574-5p | ↑ | myeloid | promotes differentiation in monocytes | [15] |
| miR-1246 | ↑ | T lymphocytes | upregulated in Tregs | [16] |
| miR-424-3p | ↑ | myeloid | involved in monocyte differentiation | [17,18] |
| miR-584-5p | ↑ | myeloid | regulated in AML CD34+ cell line | [19] |
| miR-214-3p | ↓ | T lymphocytes | upregulated in stimulated T cells and Tregs | [20,21] |
| miR-199a-3p | ↓ | myeloid | involved in leukemogenesis | [22] |
| miR-130a-3p | ↓ | T lymphocytes | upregulated in CD8 T cells inhibits CD69 | [23] |
| miR-195-5p | ↓ | myeloid | regulated in monocytes | [24] |
| miR-30c-5p | ↓ | myeloid | regulated in mDCs and pDCs | [25] |
| let-7c-5p | ↓ | myeloid | promotes M2 polarization | [26,27] |
| miR-23a-3p | ↓ | T lymphocytes; myeloid | represses CTL activity; regulated in mDCs | [25,28] |
| miR-99a-5p | ↓ | B lymphocytes | involved in B cell differentiation | [29] |
| miR-199a-5p | ↓ | T lymphocytes | overexpressed in T cells vs ALCL ALK+ | [30] |
| miR-126-3p | ↓ | T lymphocytes | downregulated in activated T cells | [31] |
| miR-193b-3p | ↓ | T lymphocytes | involved in T cell maturation | [32] |
| let-7e-5p | ↓ | myeloid | regulated in macrophage upon TLR4 | [33] |
| miR-365a-3p | ↓ | T lymphocytes | upregulated in T activation | [31] |
| miR-10b-5p | ↓ | myeloid | upregulated in CD34+ in myelofibrosis | [34] |
| miR-139-5p | ↓ | T lymphocytes | regulated in CTLs | [35] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallacchi, V.; Camisaschi, C.; Dugo, M.; Vergani, E.; Deho, P.; Gualeni, A.; Huber, V.; Gloghini, A.; Maurichi, A.; Santinami, M.; et al. microRNA Expression in Sentinel Nodes from Progressing Melanoma Patients Identifies Networks Associated with Dysfunctional Immune Response. Genes 2016, 7, 124. https://doi.org/10.3390/genes7120124
Vallacchi V, Camisaschi C, Dugo M, Vergani E, Deho P, Gualeni A, Huber V, Gloghini A, Maurichi A, Santinami M, et al. microRNA Expression in Sentinel Nodes from Progressing Melanoma Patients Identifies Networks Associated with Dysfunctional Immune Response. Genes. 2016; 7(12):124. https://doi.org/10.3390/genes7120124
Chicago/Turabian StyleVallacchi, Viviana, Chiara Camisaschi, Matteo Dugo, Elisabetta Vergani, Paola Deho, Ambra Gualeni, Veronica Huber, Annunziata Gloghini, Andrea Maurichi, Mario Santinami, and et al. 2016. "microRNA Expression in Sentinel Nodes from Progressing Melanoma Patients Identifies Networks Associated with Dysfunctional Immune Response" Genes 7, no. 12: 124. https://doi.org/10.3390/genes7120124
APA StyleVallacchi, V., Camisaschi, C., Dugo, M., Vergani, E., Deho, P., Gualeni, A., Huber, V., Gloghini, A., Maurichi, A., Santinami, M., Sensi, M., Castelli, C., Rivoltini, L., & Rodolfo, M. (2016). microRNA Expression in Sentinel Nodes from Progressing Melanoma Patients Identifies Networks Associated with Dysfunctional Immune Response. Genes, 7(12), 124. https://doi.org/10.3390/genes7120124