
Evaluation of Methylation Biomarkers for Detection of Circulating Tumor DNA and Application to Colorectal Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Samples
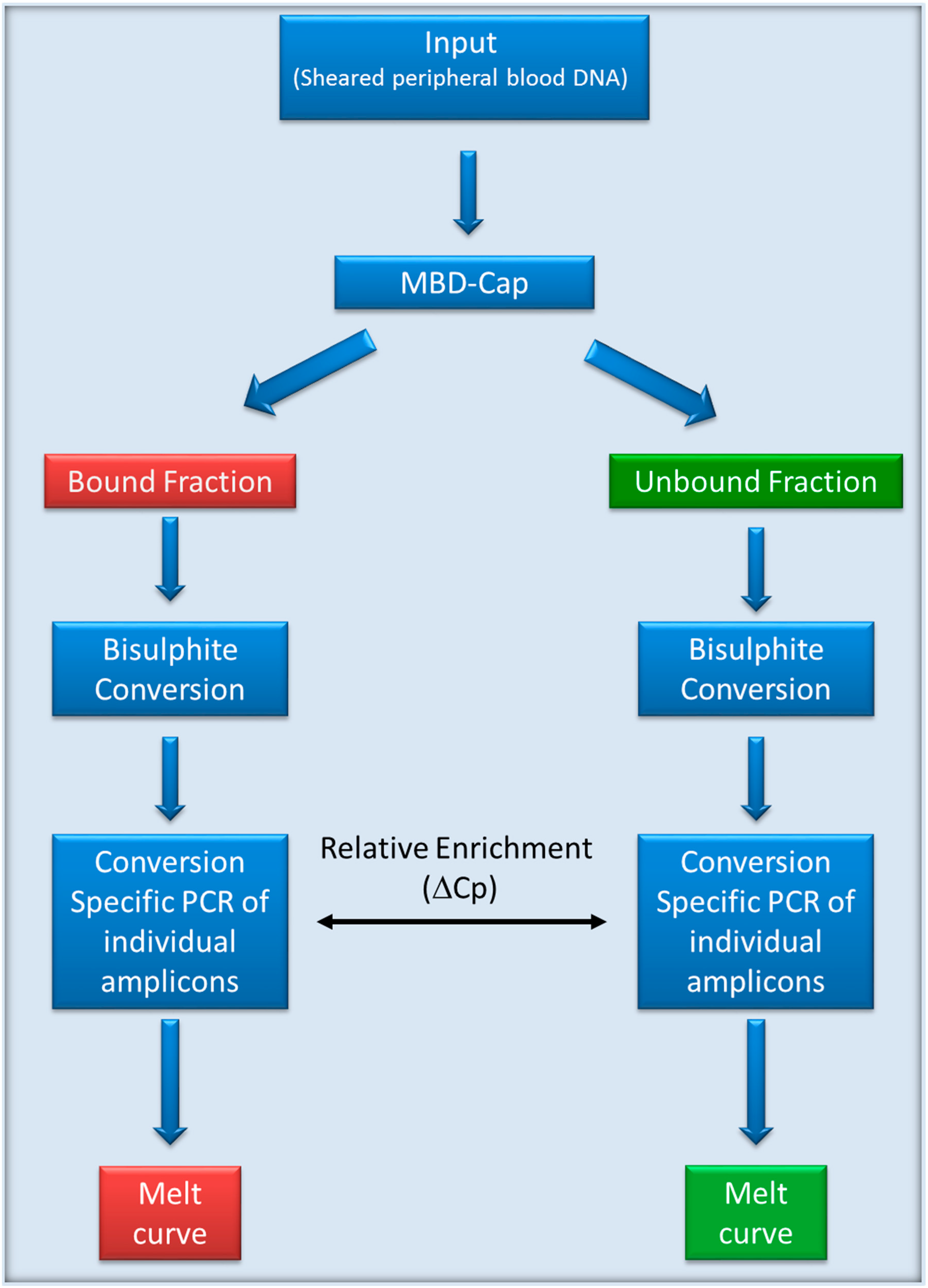
2.2. MethylMiner Enrichment of Genomic DNA
2.3. Real-time PCR Quantification
2.4. Methylation Specific PCR Assays
3. Results
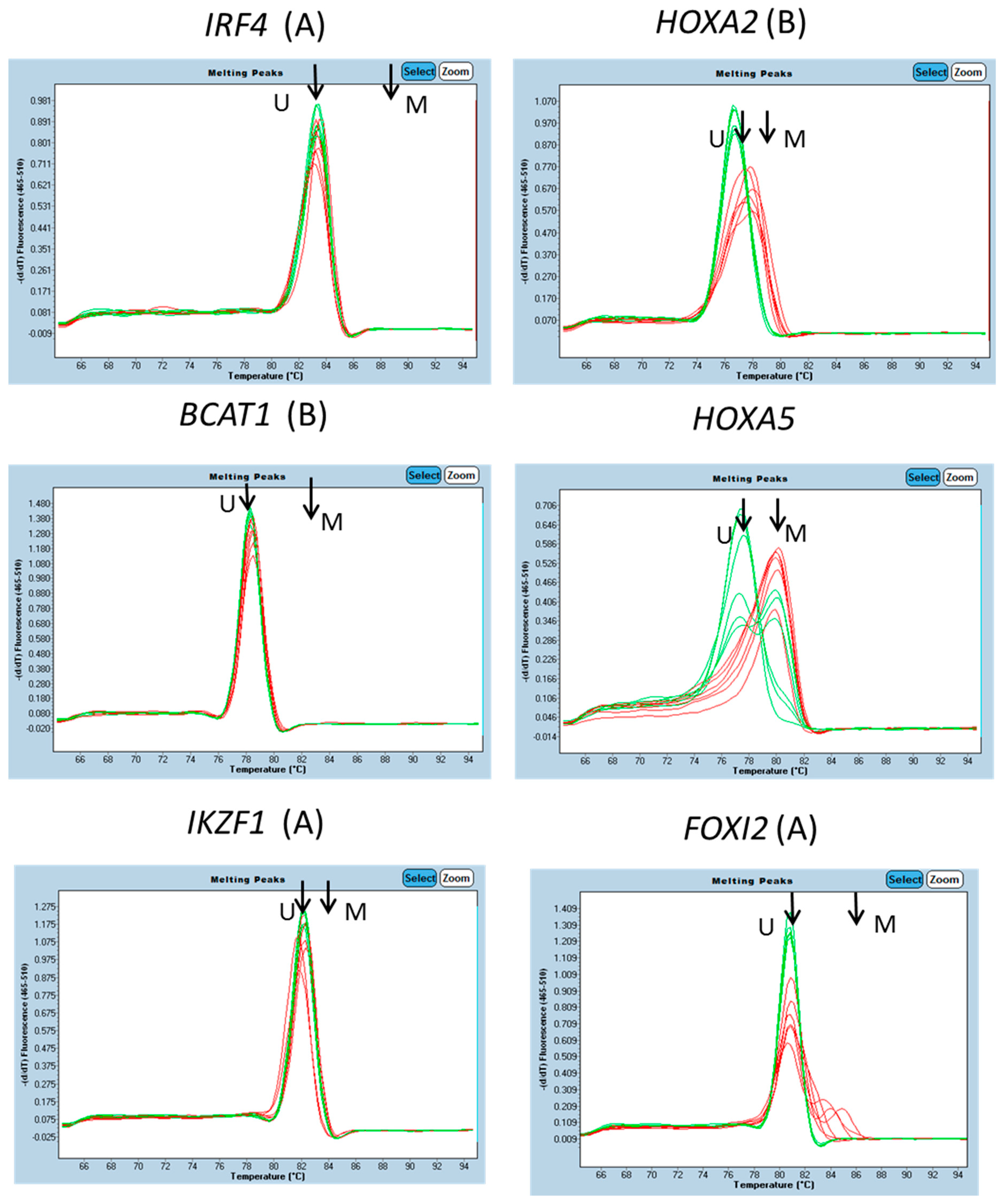
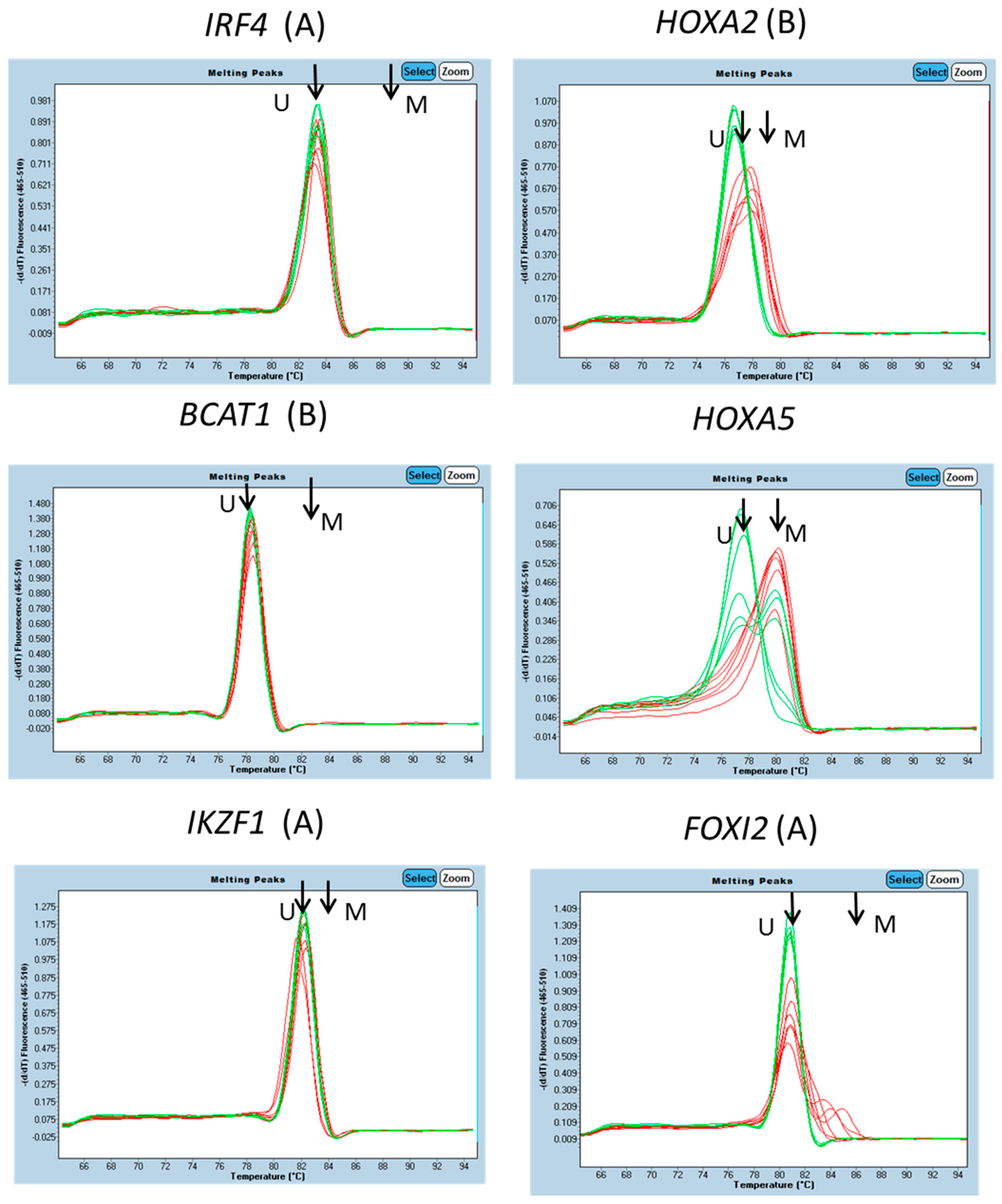
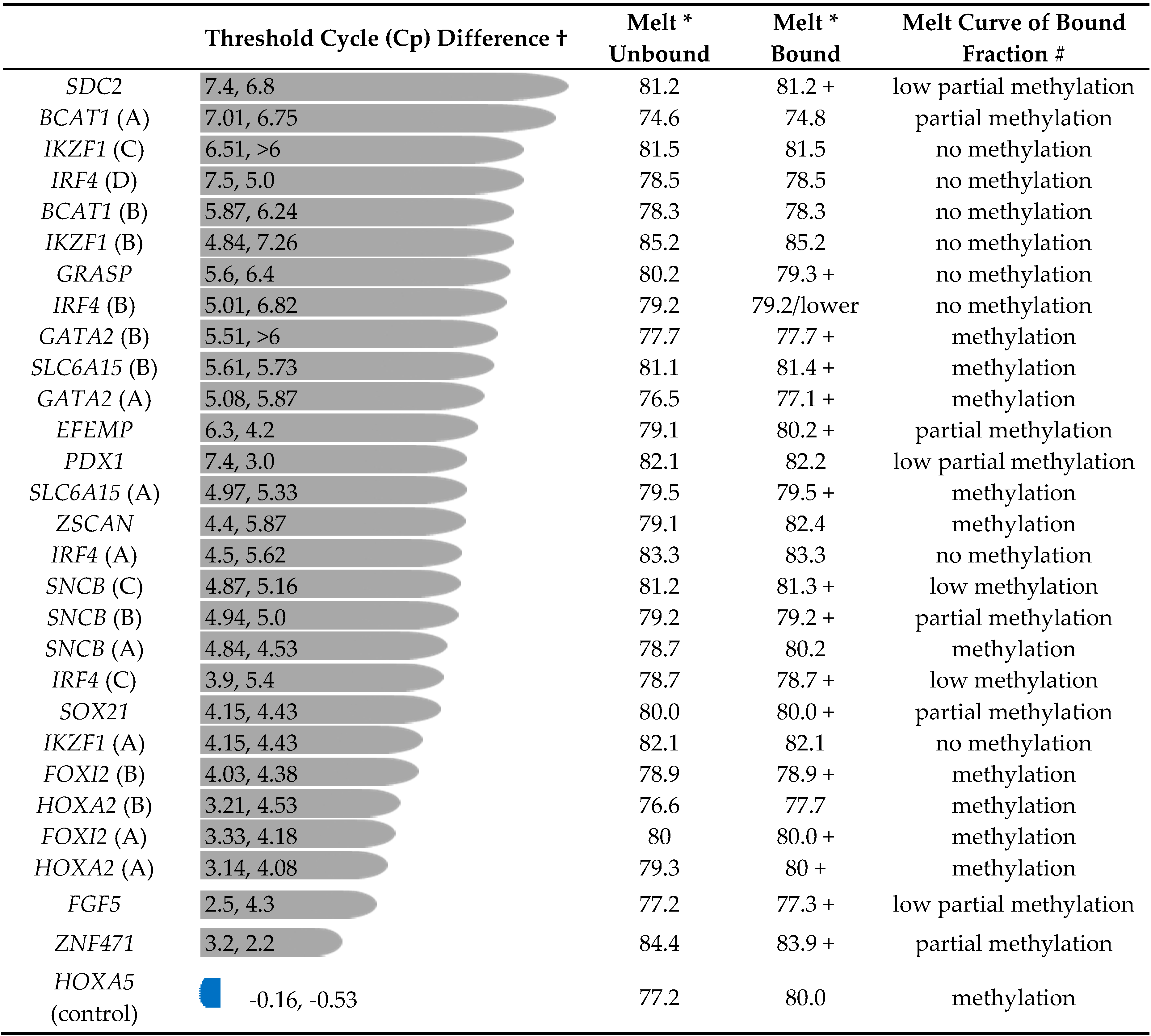
3.1. Characterisation of Methylation in WBC DNA
3.2. Evaluation of Selected Candidates in Human Plasma
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bordi, P.; Del Re, M.; Danesi, R.; Tiseo, M. Circulating DNA in diagnosis and monitoring EGFR gene mutations in advanced non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 584–597. [Google Scholar] [PubMed]
- El Messaoudi, S.; Mouliere, F.; Du Manoir, S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.; et al. Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, X.-H.; Yuan, J.-Q.; Yang, Z.-Y.; Mao, C.; Dong, X.-M.; Tang, J.-L.; Wang, S.-Y. Colorectal cancer: Using blood samples and tumor tissue to detect K-ras mutations. Expert Rev. Anticancer Ther. 2015, 15, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska-Swojak, M.; Handschuh, L.; Wojciechowski, P.; Goralski, M.; Tomaszewski, K.; Kazmierczak, M.; Lewandowski, K.; Komarnicki, M.; Blazewicz, J.; Figlerowicz, M.; et al. Simultaneous detection of mutations and copy number variation of NPM1 in the acute myeloid leukemia using multiplex ligation-dependent probe amplification. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2016, 786, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine methylation of regulatory sequences near the π-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef] [PubMed]
- Millar, D.S.; Ow, K.K.; Paul, C.L.; Russell, P.J.; Molloy, P.L.; Clark, S.J. Detailed methylation analysis of the glutathione S-transferase π (GSTP1) gene in prostate cancer. Oncogene 1999, 18, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Liebenberg, V.; Dietrich, D.; Schlegel, T.; Kneip, C.; Seegebarth, A.; Flemming, N.; Seemann, S.; Distler, J.; Lewin, J.; et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 2010, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- DeVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Gruetzmann, R.; Pilarsky, C.; Habermann, J.K.; Fleshner, P.; et al. Circulating methylated septin 9 DNA in plasma is a biomarker for colorectal cancer. Gastroenterology 2009, 136, A623. [Google Scholar] [CrossRef]
- Lange, C.P.E.; Campan, M.; Hinoue, T.; Schmitz, R.F.; van der Meulen-de Jong, A.E.; Slingerland, H.; Kok, P.J.M.J.; van Dijk, C.M.; Weisenberger, D.J.; Shen, H.; et al. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS ONE 2012, 7, e50266. [Google Scholar] [CrossRef] [PubMed]
- Melotte, V.; Lentjes, M.H.F.M.; van den Bosch, S.M.; Hellebrekers, D.M.E.I.; de hoon, J.P.J.; Wouters, K.A.D.; Daenen, K.L.J.; Partouns-Hendriks, I.E.J.M.; Stessels, F.; Louwagie, J.; et al. N-Myc downstream-regulated gene 4 (NDRG4): A candidate tumor suppressor gene and potential biomarker for colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.M.; Ross, J.P.; Drew, H.R.; Ho, T.; Brown, G.S.; Saunders, N.F.W.; Duesing, K.R.; Buckley, M.J.; Dunne, R.; Beetson, I.; et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.J.; Cooper, W.N.; Grundy, R.G.; Caldwell, G.; Jones, C.; Wadey, R.B.; Morton, D.; Schofield, P.N.; Reik, W.; Latif, F.; et al. Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene 2002, 21, 7277–7282. [Google Scholar] [CrossRef] [PubMed]
- Avraham, A.; Uhlmann, R.; Shperber, A.; Birnbaum, M.; Sandbank, J.; Sella, A.; Sukumar, S.; Evron, E. Serum DNA methylation for monitoring response to neoadjuvant chemotherapy in breast cancer patients. Int. J. Cancer 2012, 131, E1166–E1172. [Google Scholar] [CrossRef] [PubMed]
- Brait, M.; Sidransky, D. Cancer epigenetics: Above and beyond. Toxicol. Mech. Methods 2011, 21, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Qu, W.; Devaney, J.; Paul, C.; Castillo, L.; Wykes, R.J.; Chatfield, M.D.; Boyer, M.J.; Stockler, M.R.; Marx, G.; et al. Methylated glutathione S-transferase 1 (MGSTP1) is a potential plasma free DNA epigenetic marker of prognosis and response to chemotherapy in castrate-resistant prostate cancer. Br. J. Cancer 2014, 111, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Kneip, C.; Raji, O.; Lloglou, T.; Seegebarth, A.; Schlegel, T.; Flemming, N.; Rausch, S.; Distler, J.; Fleischhacker, M.; et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int. J. Oncol. 2012, 40, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Fantony, J.J.; Abern, M.R.; Gopalakrishna, A.; Owusu, R.; Jack Tay, K.; Lance, R.S.; Inman, B.A. Multi-institutional external validation of urinary TWIST1 and NID2 methylation as a diagnostic test for bladder cancer. Urol. Oncol. 2015, 33, 387.e1–387.e6. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Lin, C.Y.; Yang, S.F.; Ho, C.M.; Chang, J.G. Analysing the mutational status of adenomatous polyposis coli (APC) gene in breast cancer. Cancer Cell Int. 2016, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.P.E.; Laird, P.W. Clinical applications of DNA methylation biomarkers in colorectal cancer. Epigenomics 2013, 5, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Raynor, M.P.; Candiloro, I.; Dobrovic, A. Methylation profiling of normal individuals reveals mosaic promoter methylation of cancer-associated genes. Oncotarget 2012, 3, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.K.; Baker, R.T.; McEvoy, A.; Murray, D.H.; Thomas, M.; Molloy, P.L.; Mitchell, S.; Lockett, T.; Young, G.P.; LaPointe, L.C. A two-gene blood test for methylated DNA sensitive for colorectal cancer. PLoS ONE 2015, 10, e0125041. [Google Scholar] [CrossRef] [PubMed]
- Symonds, E.L.; Pedersen, S.K.; Baker, R.T.; Murray, D.H.; Gaur, S.; Cole, S.R.; Gopalsamy, G.; Mangira, D.; LaPointe, L.C.; Young, G.P. A blood test for methylated BCAT1 and IKZF1 vs. a fecal immunochemical test for detection of colorectal neoplasia. Clin. Transl. Gastroenterol. 2016, 7, e137. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.K.; Symonds, E.L.; Baker, R.T.; Murray, D.H.; McEvoy, A.; Van Doorn, S.C.; Mundt, M.W.; Cole, S.R.; Gopalsamy, G.; Mangira, D.; et al. Evaluation of an assay for methylated BCAT1 and IKZF1 in plasma for detection of colorectal neoplasia. BMC Cancer 2015, 15, 654. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Byrd, D.R.; Compton, C.C.; Fritz, A.G.; Greene, F.L.; Trotti, A. AJCC Cancer Staging Manual, 7th ed.; Springer: New York, NY, USA, 2010. [Google Scholar]
- Kulis, M.; Merkel, A.; Heath, S.; Queirós, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Oakes, C.C.; Seifert, M.; Assenov, Y.; Gu, L.; Przekopowitz, M.; Ruppert, A.; Wang, Q.; Imbusch, C.D.; Serva, A.; Koser, S.D.; et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 2016, 48, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.N.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Hansen, J.W.; Kristensen, S.S.; Tholstrup, D.; Harsløf, L.B.; Pedersen, O.B.; De Nully Brown, P.; Grønbæk, K. Aberrant methylation of cell-free circulating DNA in plasma predicts poor outcome in diffuse large B cell lymphoma. Clin. Epigenet. 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-wide identification and validation of a novel methylation biomarker, SDC2, for blood-based detection of colorectal cancer. J. Mol. Diagn. 2013, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Lai, S.C.; Xu, Z.P.; Wang, L.J. Noninvasive DNA methylation biomarkers in colorectal cancer: A systematic review. J. Dig. Dis. 2015, 16, 699–712. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Cancer | Adenoma | Normal | ||||
|---|---|---|---|---|---|---|
| Number Tested | Number (%) Positive | Number Tested | Number (%) Positive | Number Tested | Number (%) Positive | |
| BCAT1 | 74 | 48 (64.9) | 33 | 4 (12.1) | 144 | 5 (3.5) |
| FGF5 | 20 | 17 (85) | 40 | 13 (32.5) | 40 | 7 (17.5) |
| GRASP | 44 | 24 (54.5) | 44 | 6 (13.6) | 44 | 3 (6.8) |
| IKZF1 | 74 | 50 (67.6) | 33 | 8 (24.2) | 144 | 7 (4.9) |
| IRF4 | 22 | 13 (59.1) | 21 | 2 (9.5) | 24 | 1 (4.2) |
| PDX1 | 20 | 9 (45) | 20 | 4 (20) | 20 | 6 (30) |
| SDC2 | 44 | 26 (59.1) | 44 | 6 (13.6) | 44 | 7 (15.9) |
| SEPT9 | 44 | 26 (59.1) | 44 | 2 (4.5) | 44 | 2 (4.5) |
| SOX21 | 20 | 17 (85) | 17 | 9 (52.9) | 20 | 10 (50) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitchell, S.M.; Ho, T.; Brown, G.S.; Baker, R.T.; Thomas, M.L.; McEvoy, A.; Xu, Z.-Z.; Ross, J.P.; Lockett, T.J.; Young, G.P.; et al. Evaluation of Methylation Biomarkers for Detection of Circulating Tumor DNA and Application to Colorectal Cancer. Genes 2016, 7, 125. https://doi.org/10.3390/genes7120125
Mitchell SM, Ho T, Brown GS, Baker RT, Thomas ML, McEvoy A, Xu Z-Z, Ross JP, Lockett TJ, Young GP, et al. Evaluation of Methylation Biomarkers for Detection of Circulating Tumor DNA and Application to Colorectal Cancer. Genes. 2016; 7(12):125. https://doi.org/10.3390/genes7120125
Chicago/Turabian StyleMitchell, Susan M., Thu Ho, Glenn S. Brown, Rohan T. Baker, Melissa L. Thomas, Aidan McEvoy, Zheng-Zhou Xu, Jason P. Ross, Trevor J. Lockett, Graeme P. Young, and et al. 2016. "Evaluation of Methylation Biomarkers for Detection of Circulating Tumor DNA and Application to Colorectal Cancer" Genes 7, no. 12: 125. https://doi.org/10.3390/genes7120125
APA StyleMitchell, S. M., Ho, T., Brown, G. S., Baker, R. T., Thomas, M. L., McEvoy, A., Xu, Z.-Z., Ross, J. P., Lockett, T. J., Young, G. P., LaPointe, L. C., Pedersen, S. K., & Molloy, P. L. (2016). Evaluation of Methylation Biomarkers for Detection of Circulating Tumor DNA and Application to Colorectal Cancer. Genes, 7(12), 125. https://doi.org/10.3390/genes7120125