
A Review of Computational Methods for Finding Non-Coding RNA Genes


Abstract
:1. Introduction
2. Review of Computational Intelligence Techniques
2.1. Predicting Non-Coding RNAs
2.2. Finding MicroRNAs
3. Online Tools and Data Sources
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMPO Rep. 2001, 2, 957–1051. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Lv, Y.; Zhao, H.; Gong, Y.; Hu, J.; Li, F.; Xu, J.; Bai, J.; Yu, F.; Li, X. Predicting the functions of long noncoding RNAs using RNA-Seq based on Bayesian network. Biomed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Wuyts, J.; Rouzé, P.; van de Peer, Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Clote, P. RNALOSS: A web server for RNA locally optimal secondary structures. Nucleic Acids Res. 2005, 33 (Suppl. 2), W600–W604. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, D.; Di Bella, S.; Nigita, G.; Laganà, A.; Ferro, A.; Croce, C.M. Noncoding RNA: Current Deep Sequencing Data Analysis Approaches and Challenges. Hum. Mutat. 2016, 37, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z. Computational biology in microRNA. Wiley Interdiscip. Rev. RNA 2015, 6, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Chen, C.; Kaye, A.M.; Wasserman, W.W. The identification of cis-regulatory elements: A review from a machine learning perspective. Biosystems 2015, 138, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Dubchak, I.; Holbrook, S.R. A computational approach to identify genes for functional RNAs in genomic sequences. Nucleic Acids Res. 2001, 29, 3928–3938. [Google Scholar] [PubMed]
- Arslan, A.; Şen, B. Detection of non-coding RNA’s with optimized support vector machines. In Proceedings of the 2015 23nd Signal Processing and Communications Applications Conference (SIU), Malatya, Turkey, 16–19 May 2015.
- Karathanou, K.; Theofilatos, K.; Kleftogiannis, D.; Likothanassis, S.; kos, C.A.; kalidis, A.T.; Mavroudi, S. ncRNAclass: A web platform for non-coding RNA feature calculation and microRNAs and targets prediction. Int. J. Artif. Intell. Tools 2015, 24, 1540002. [Google Scholar] [CrossRef]
- Wang, C.; Ding, C.; Meraz, R.F.; Holbrook, S.R. PSoL: A positive sample only learning algorithm for finding non-coding RNA genes. Bioinformatics 2006, 22, 2590–2596. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006, 2, e29. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.C.; Stephen, S.; Engström, P.G.; Tajul-Arifin, K.; Chen, W.; Wahlestedt, C.; Lenhard, B.; Hayashizaki, Y.; Mattick, J.S. RNAdb—A comprehensive mammalian noncoding RNA database. Nucleic Acids Res. 2005, 33 (Suppl. 1), D125–D130. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bai, B.; Skogerbø, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005, 33 (Suppl. 1), D112–D115. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35 (Suppl. 2), W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Griffithsjones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- Cochrane, G.; Aldebert, P.; Althorpe, N.; Andersson, M.; Baker, W.; Baldwin, A.; Bates, K.; Bhattacharyya, S.; Browne, P.; van den Broek, A.; et al. EMBL Nucleotide Sequence Database: developments in 2005. Nucleic Acids Res. 2006, 34, D10–D15. [Google Scholar] [CrossRef] [PubMed]
- Sætrom, P.; Sneve, R.; Kristiansen, K.; Snøve, O., Jr.; Grünfeld, T.; Rognes, T.; Seeberg, E. Predicting non-coding RNA genes in Escherichia coli with boosted genetic programming. Nucleic Acids Res. 2005, 33, 3263–3270. [Google Scholar]
- Yoon, B.; Vaidyanathan, P.P. An overview of the role of context-sensitive HMMS in the prediction of NCRNA genes IEEE/SP. In Proceedings of the 13th Workshop on Statistical Signal Processing, Bordeaux, France, 17–20 July 2005.
- Saha, S.; Mitra, S.; Yadav, R.K. A Multiobjective based automatic framework for classifying cancer-microRNA biomarkers. Gene Rep. 2016, 4, 91–103. [Google Scholar] [CrossRef]
- Lee, B.; Baek, J.; Park, S.; Yoon, S. deepTarget: End-to-end learning framework for microRNA target prediction using deep recurrent neural networks. In Proceedings of the 7th ACM Conference on Bioinformatics, Computational Biology and Health Informatics (BCB), Seattle, WA, USA, 2–5 October 2016; pp. 434–442.
- Cheng, S.; Guo, M.; Wang, C.; Liu, X.; Liu, Y.; Wu, X. MiRTDL: A deep learning approach for miRNA target prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 36, 1. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Yu, Z.; Li, B.; Gu, F.; Pan, Y. A Comprehensive review of emerging computational methods for gene identification. J. Inf. Process. Syst. 2016, 12, 1–34. [Google Scholar]
- Rahman, E.; Islam, R.; Islam, S.; Mondal, S.I.; Amin, R. MiRANN: A reliable approach for improved classification of precursor microRNA using Artificial Neural Network model. Genomics 2012, 99, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, D.; Chen, Y. Prediction of pre-miRNA with multiple stem-loops using feedforward neural network. Intell. Comput. Theor. Methodol. 2015, 9226, 554–562. [Google Scholar]
- Tran, T.T.; Zhou, F.; Marshburn, S.; Stead, M.; Kushner, S.R.; Xu, Y. De novo computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics 2009, 25, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L. Statistical study on disease-related ncRNAs using Z-curve method. In Proceedings of the 2009 International Conference on Computational Intelligence and Natural Computing, Wuhan, China, 6–7 June 2009; pp. 503–506.
- Smith, S.F. A Genetic algorithms approach to non-coding RNA gene searches. In Proceedings of the 2006 IEEE Mountain Workshop on Adaptive and Learning Systems, Logan, UT, USA, 24–26 July 2006.
- Smith, S.F. Covariance Searches for ncRNA gene finding. In Proceedings of the 2006 IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology, Toronto, ON, Canada, 28–29 September 2006.
- Tseng, H.; Weinberg, Z.; Gore, J.; Breaker, R.R.; Ruzzo, W.L. Finding non-coding RNAs through genome-scale clustering. J. Bioinform. Comput. Biol. 2009, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, F.; He, T.; Liu, G.; Li, Y.; Zhang, X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinform. 2005, 6, 310. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Eddy, S.R. Computational identification of non-coding RNAs in Saccharomyces cerevisiae by comparative genomics. Nucleic Acids Res. 2003, 31, 4119–4128. [Google Scholar] [CrossRef] [PubMed]
- Numata, K.; Kanai, A.; Saito, R.; Kondo, S.; Adachi, J.; Wilming, L.G.; Hume, D.A.; Hayashizaki, Y.; Tomita, M.; RIKEN GER Group; GSL Members. Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. Genome Res. 2003, 13, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P. Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 2002, 30, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Eddy, S.R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinform. 2001, 2, 8. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Wu, H.; Wang, W.; Ma, W.; Sun, X.; Lu, Z. MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007, 35, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Loong, K.; Mishra, S. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics 2007, 23, 1321–1330. [Google Scholar]
- Sewer, A.; Paul, N.; Landgraf, P.; Aravin, A.; Pfeffer, S.; Brownstein, M.J.; Tuschl, T.; van Nimwegen, E.; Zavolan, M. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinform. 2005, 6, 267–282. [Google Scholar] [PubMed]
- Batuwita, R.; Palade, V. microPred: effective classification of pre-miRNAs for human miRNA gene prediction. Bioinformatics 2009, 25, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, F.; He, T.; Liu, G.P.; Li, Y.; Zhang, X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinform. 2005, 6, 310. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.R.; Eddy, S.R. RSEARCH: Finding homologs of single structured RNA sequences. BMC Bioinform. 2003, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, I.; Saito, C.; Wakamoto, A.; Tomaru, Y.; Arakawa, T.; Hayashizaki, Y.; Okazaki, Y. EICO (Expression-based Imprint Candidate Organizer): Finding disease-related imprinted genes. Nucleic Acids Res. 2004, 32 (Suppl. 1), D548–D551. [Google Scholar]
- Zhang, S.; Haas, B.; Eskin, E.; Bafna, V. Searching genomes for noncoding RNA using FastR. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005, 2, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Fritah, S.; Niclou, S.P.; Azuaje, F. Databases for lncRNAs: A comparative evaluation of emerging tools. RNA 2014, 2011, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.S.; Maiti, S. Noncoding RNAs and their annotation using metagenomics algorithms. Wiley Interdiscip. Rev. 2015, 5, 1–20. [Google Scholar] [CrossRef]
- Yu, N.; Cho, K.H.; Cheng, Q.; Tesorero, R.A. A hybrid computational approach for the prediction of small non-coding RNAs from genome sequences. In Proceedings of the International Conference on Computational Science and Engineering, Vancouver, BC, Canada, 29–31 August 2009.
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Vucic, E.A.; Enfield, K.S.S.; Stewart, G.L.; Lonergan, K.M.; Kennett, J.Y.; Becker-Santos, D.D.; MacAulay, C.E.; Lam, S.; Brown, C.J.; Lam, W.L. Human cancer long non-coding RNA transcriptomes. PLoS ONE 2011, 6, e25915. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Gao, L.; Wang, Y.; Chiu, D.K.; Wang, T.; Deng, Y. Advances in long noncoding RNAs: Identification, structure prediction and function annotation. Brief. Funct. Genom. 2016, 15, 38–46. [Google Scholar]
- Zou, Q.; Guo, M.; Liu, Y.; Xing, Z. A novel comparative sequence analysis method for ncRNA secondary structure prediction without multiple sequence alignment. In Proceedings of the 2008 Fourth International Conference on Natural Computation, Jinan, China, 18–20 October 2008.
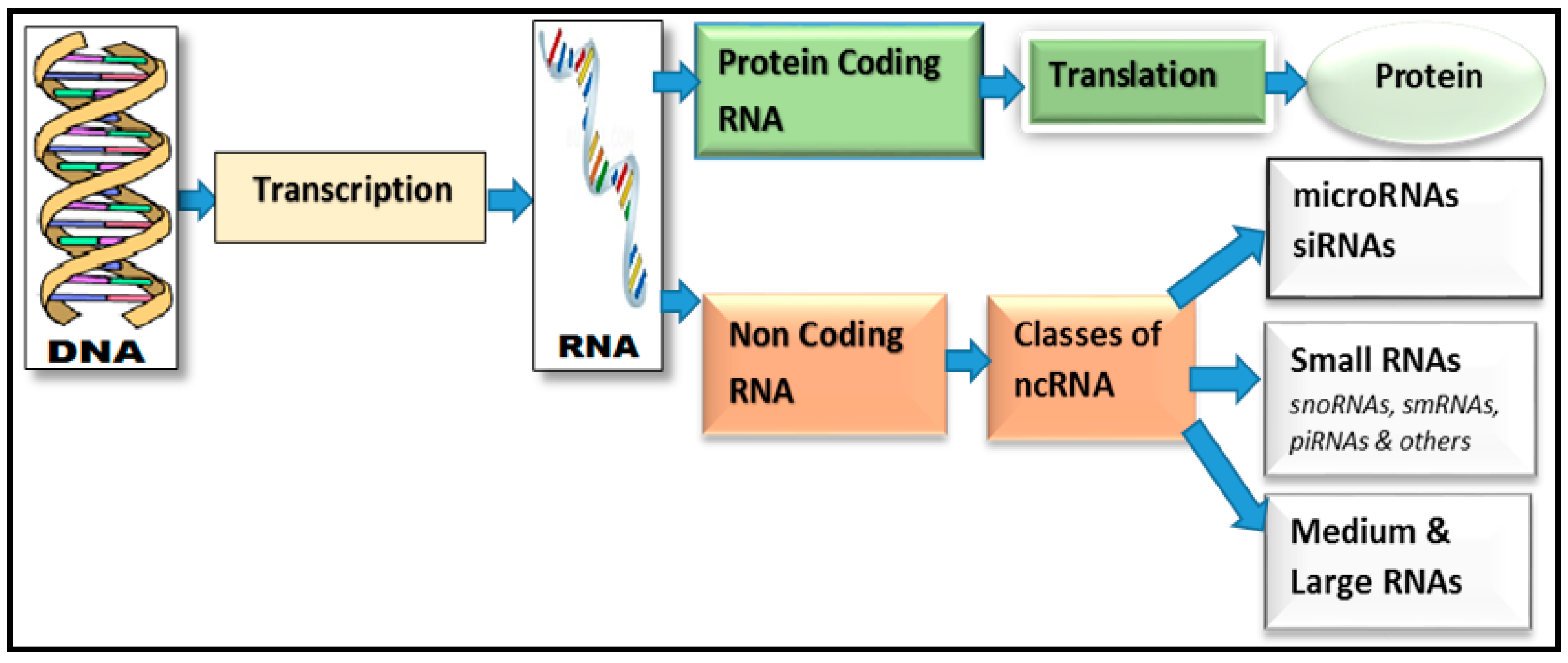

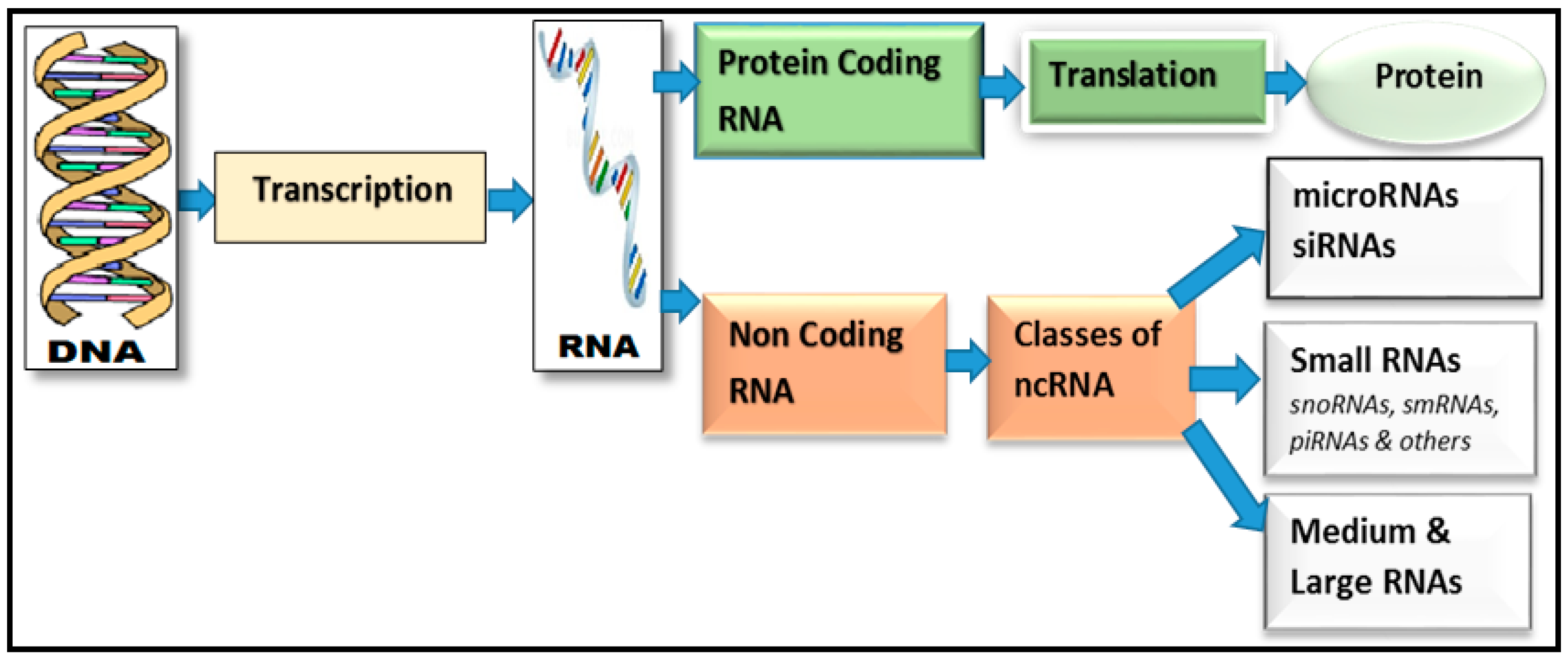{kind=link}
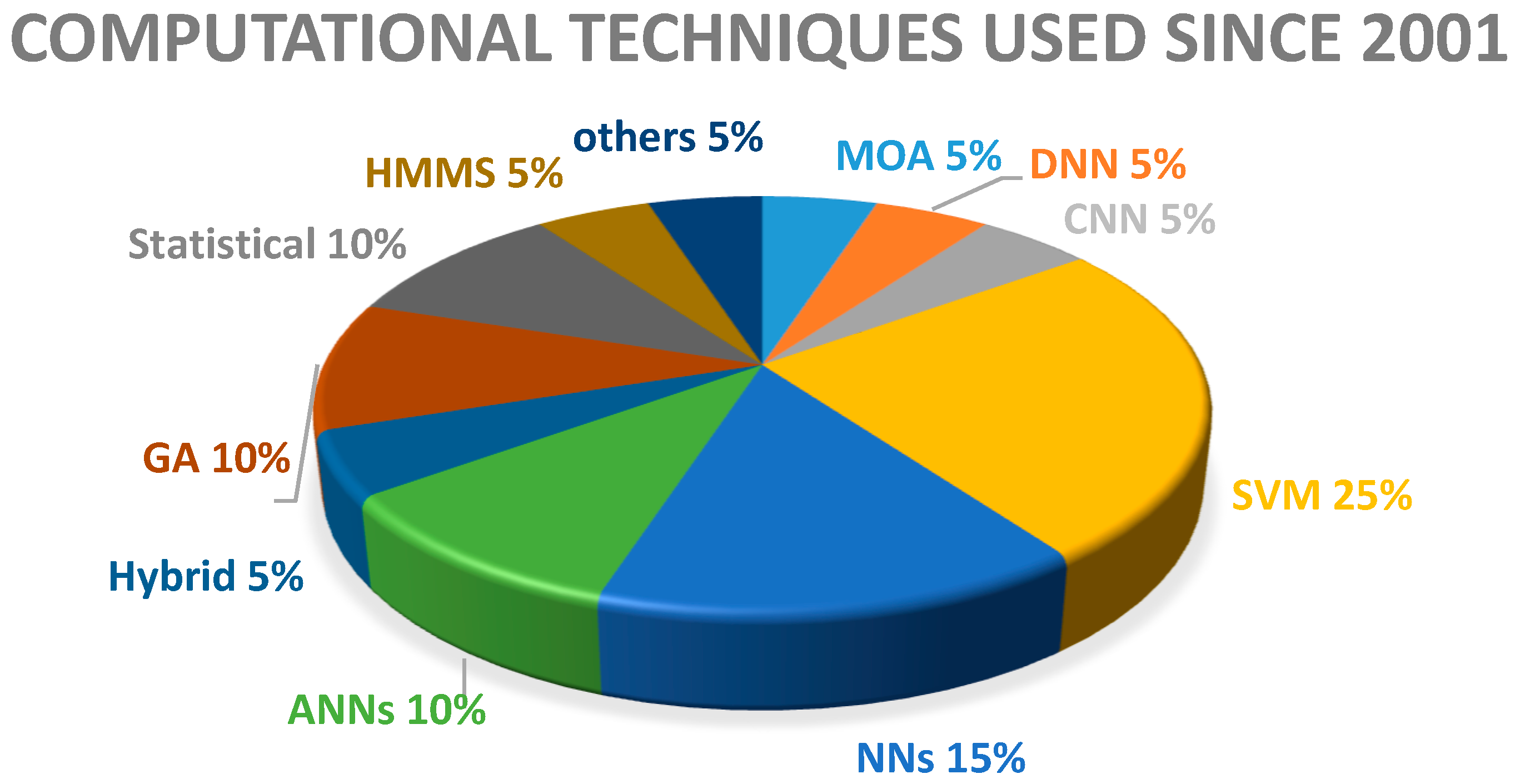
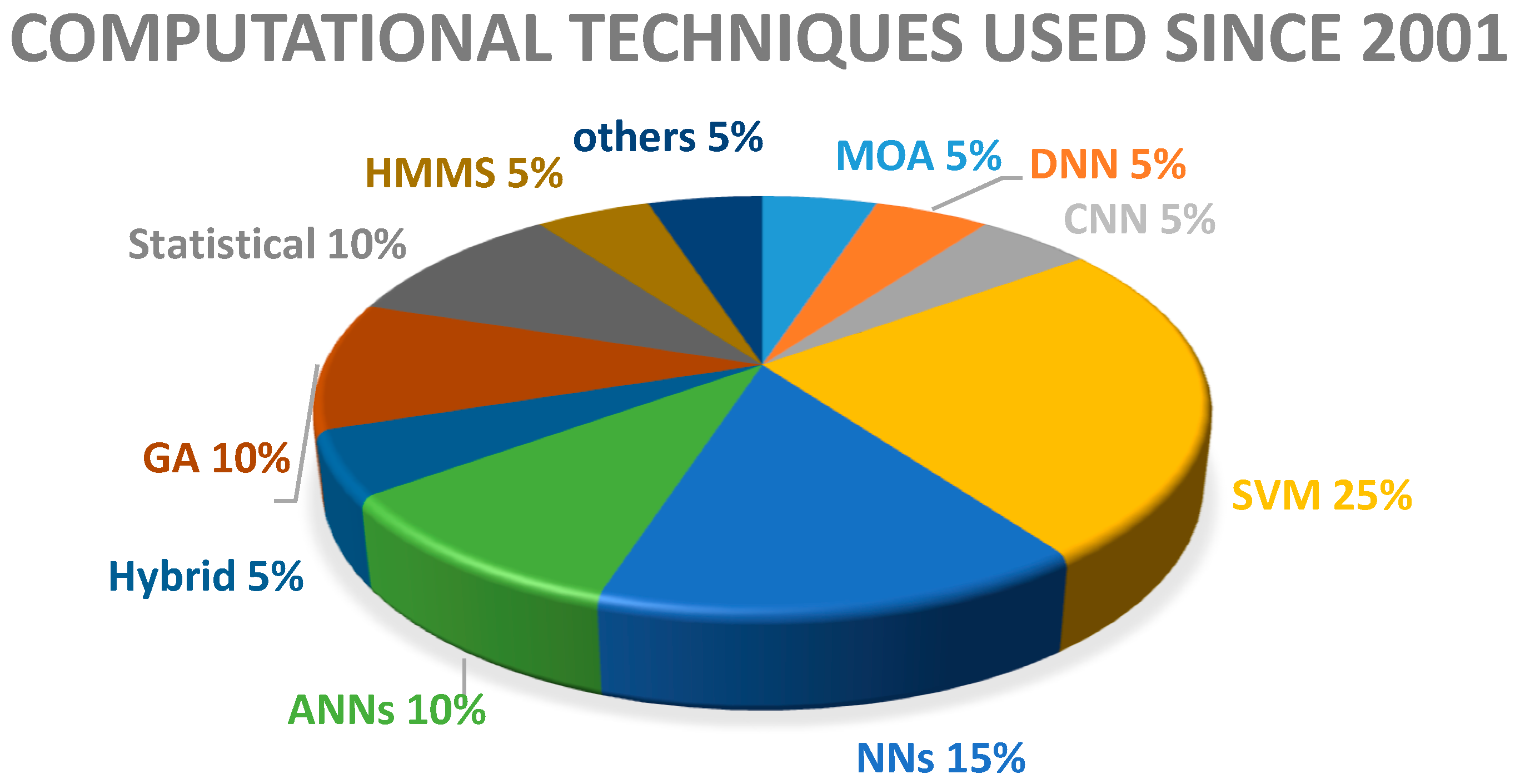{kind=link}
| Cited | Approach | 4 CMP | Results (%) | Methodology and Online Resource Tools |
|---|---|---|---|---|
| [13] | A computational approach to identify genes for functional RNAs in genomic sequences. | √ | 2 S: 90%, 3 P: 99% | NN and SVM. Online tool unavailable. |
| [14] | To detect ncRNA sequences. | × | −−−−−− | The support vector machine (SVM) algorithm was implemented in graphical processing units (GPUs) based parallel technology. Online tool unavailable. |
| [15] | To differentiate between well-known classes and target predicted classes of messenger RNA (mRNA). | √ | −−−−−− | A new web-based interface was developed to detect ncRNAs. Available at http://biotools.ceid.upatras.gr/ncrnaclass/. |
| [16] | To identify ncRNA a positive sample only learning algorithm is introduced. | × | 1 A: 80% | The SVM used as the core learning machine assessed by 5-fold-validation in recovery of known ncRNA. Data available online at (http://bioinformatics.oxfordjournals.org/content/22/21/2590/suppl/DC1) |
| [17] | To introduce a method to differentiate between coding or non-coding RNA. | × | 3 P: 97%, 2 S: 98% | Supervised machine learning SVM is used to classify transcripts according to features they would have if transcripts coded for proteins. Online data source of mRNA at: RNAdb (http://research.imb.uq.edu.au/rnadb). |
| [20] | To identify ncRNA using six features extracted from transcript’s nucleotide sequence. | × | −−−−−− | SVM (coding potential calculator ((CPC)) to identify ncRNA using six features extracted from transcript’s nucleotide sequence. Dataset used Rfam and RNAdb for noncoding and EMBL CDS for coding. Online web-based interface available of CPC at http://cpc.cbi.pku.edu.cn. |
| [23] | The prediction of ncRNA genes using boosted genetic programming. | × | 1 A: 80% | The GA and 10-fold cross validation was used to train and test the learning machine. Online tool unavailable. |
| [25] | To classify micro RNAs (miRNAs) and to differentiate between normal and tumor tissues. | √ | −−−−−− | A multi-objective algorithm was developed by using four classifiers such as random tree (RT), random forest (RF), sequential minimal optimization (SMO) and logistic regression (LR). |
| [26] | To automatically predict miRNA target. | √ | F-measure: 0.95 | The deep neural-network (DNN) was utilized to increase F-measure by 25% for prediction of miRNA targets. Available at (http://data.snu.ac.kr/pub/deepTarget) |
| [27] | To predict miRNAs targets. | × | 1 A: 90%, 2 S: 88%, 3 P: 94% | Contrast relaxing and convolutional neural network (CNN) methods. Online tool unavailable. |
| [28] | To predict new miRNA, known as pre-miRNAs. | × | 1 A: 99.9%, 2 S: 99.8%, 3 P: 100% | A neural networks (NNs) classifier was used to predict miRNA. Online tool unavailable. |
| [29] | To improve the performance and to predict the regulation of miRNA. | × | −−−−−−−− | The authors utilized a NNs classifier to predict miRNA. Online tool unavailable. |
| [30] | To predict a real pre-miRNA or a pseudo pre-miRNA. | √ | 1 S: 97.40%, 2 P: 95.85% | The authors utilized a multilayer artificial neural network (ANN) classifier. Online tool unavailable. |
| [31] | A de novo prediction algorithm to identify ncRNA using features derived from sequence and structure of known ncRNA. | × | 2 S: 68%, 3 P: 70%, 1 A: 70% | NN-based meta-learner de novo predictor using folding, ensemble, and structure-based features. Online data and program found at: http://csbl.bmb.uga.edu/publications/materials/tran/ |
| [32] | The 15 disease related ncRNAs sequences are utilized from the ncRNAs with Alzheimer disease. | × | −−−−−− | From the NONCODE database [19], 15 disease related ncRNA sequences were selected for mapping and comparison. The ncRNA sequences in the cellular process and the base content in these sequences have almost the same Z-curves even though they are coming from different organisms. Online tool unavailable. |
| [33] | To identify ncRNA genes using a genetic algorithm (GA). | × | −−−−−− | The observed sequence in real sequence data is used to motivate the use of GAs to quickly reject regions of the search space of ncRNAs. Online tool unavailable. |
| [34] | To identify ncRNA using covariance searching. | × | −−−−−− | The covariance models for ncRNA gene finding is extremely powerful and also extremely computationally demanding. Online tool unavailable. |
| [35] | A comparative genomic approach is used to detect ncRNA. | × | −−−−−− | Developed an efficient clustering method for finding potential ncRNAs in bacteria by clustering genomic sequences. Online tool unavailable. |
| [36] | To identify real and pseudo miRNA using SVM with features that are present in local structure-sequence. | × | 1 A: 90% | A method to classify real and pseudo miRNA by applying SVM using local structure sequence features. Online tool unavailable. |
| [37] | Computational identification of ncRNAs in Saccharomyces cerevisiae by comparative genomics. | × | −−−−−− | Computational screen followed by Northern blot and transcript sequencing. Online tool unavailable. Data set is available only at: http://genome.cshlp.org/content/13/6b/1301/suppl/DC1. |
| [38] | Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. | × | −−−−−− | The authors identified nine ncRNAs. Online tool unavailable. Data set is available only at: http://genome.cshlp.org/content/13/6b/1301/suppl/DC1. |
| [39] | The 19 candidate ncRNAs were identified including one with significant homology. | × | −−−−−− | The author used base-composition statistics method to find variety of ncRNAs. Online tool unavailable. |
| [40] | ncRNA gene detection using comparative sequence analysis. | √ | 2 S: 97.3%, 3 P: 100% | Comparative sequence analysis algorithm with “pair grammars” based on stochastic and hidden Markov models (HMM). Online tool unavailable. |
| Year | Computational Intelligence |
|---|---|
| 2016 | Multi classifiers (RT, RF, SMO) and Logic Regression LRDNN |
| 2015 | CNN, SVM, NN |
| 2012 | NN |
| 2009 | ANN, De novo NN, Hybrid Methods (HMs) |
| 2008 | Z-curve, GA |
| 2007 | SVM-Coding |
| 2006 | SVM and Covariance model parameter estimation |
| 2005 | GA, SVM, HMMs |
| 2002 | Local base-composition statistics |
| 2001 | Single-hidden layer NNs and SVMs, Comparative sequence analysis algorithm based on HMMs |
| Cited | Databases | Web-Links |
|---|---|---|
| [9] | RNALOSS | http://clavius.bc.edu/~clotelab/RNALOSS |
| [18] | RNAdb | http://research.imb.uq.edu.au/RNAdb |
| [19] | NONCODE | http://noncode.bioinfo.org.cn |
| [21] | Rfam | http://www.sanger.ac.uk/science/tools/rfam |
| [49] | RSEARCH | http://www.yeastgenome.org |
| ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/SEQUENCES | ||
| [50] | EICO | http://fantom2.gsc.riken.jp/EICODB/ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, Q.; Raza, S.M.; Biyabani, A.A.; Jaffar, M.A. A Review of Computational Methods for Finding Non-Coding RNA Genes. Genes 2016, 7, 113. https://doi.org/10.3390/genes7120113
Abbas Q, Raza SM, Biyabani AA, Jaffar MA. A Review of Computational Methods for Finding Non-Coding RNA Genes. Genes. 2016; 7(12):113. https://doi.org/10.3390/genes7120113
Chicago/Turabian StyleAbbas, Qaisar, Syed Mansoor Raza, Azizuddin Ahmed Biyabani, and Muhammad Arfan Jaffar. 2016. "A Review of Computational Methods for Finding Non-Coding RNA Genes" Genes 7, no. 12: 113. https://doi.org/10.3390/genes7120113
APA StyleAbbas, Q., Raza, S. M., Biyabani, A. A., & Jaffar, M. A. (2016). A Review of Computational Methods for Finding Non-Coding RNA Genes. Genes, 7(12), 113. https://doi.org/10.3390/genes7120113