
Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and RNA Isolation
2.3. cDNA Synthesis and HiSeq 2500 Sequencing
2.4. De Novo Assembly and Assessment of De Novo Assemblies
2.5. Transcriptome Annotation
2.6. Identification of SSRs
3. Results
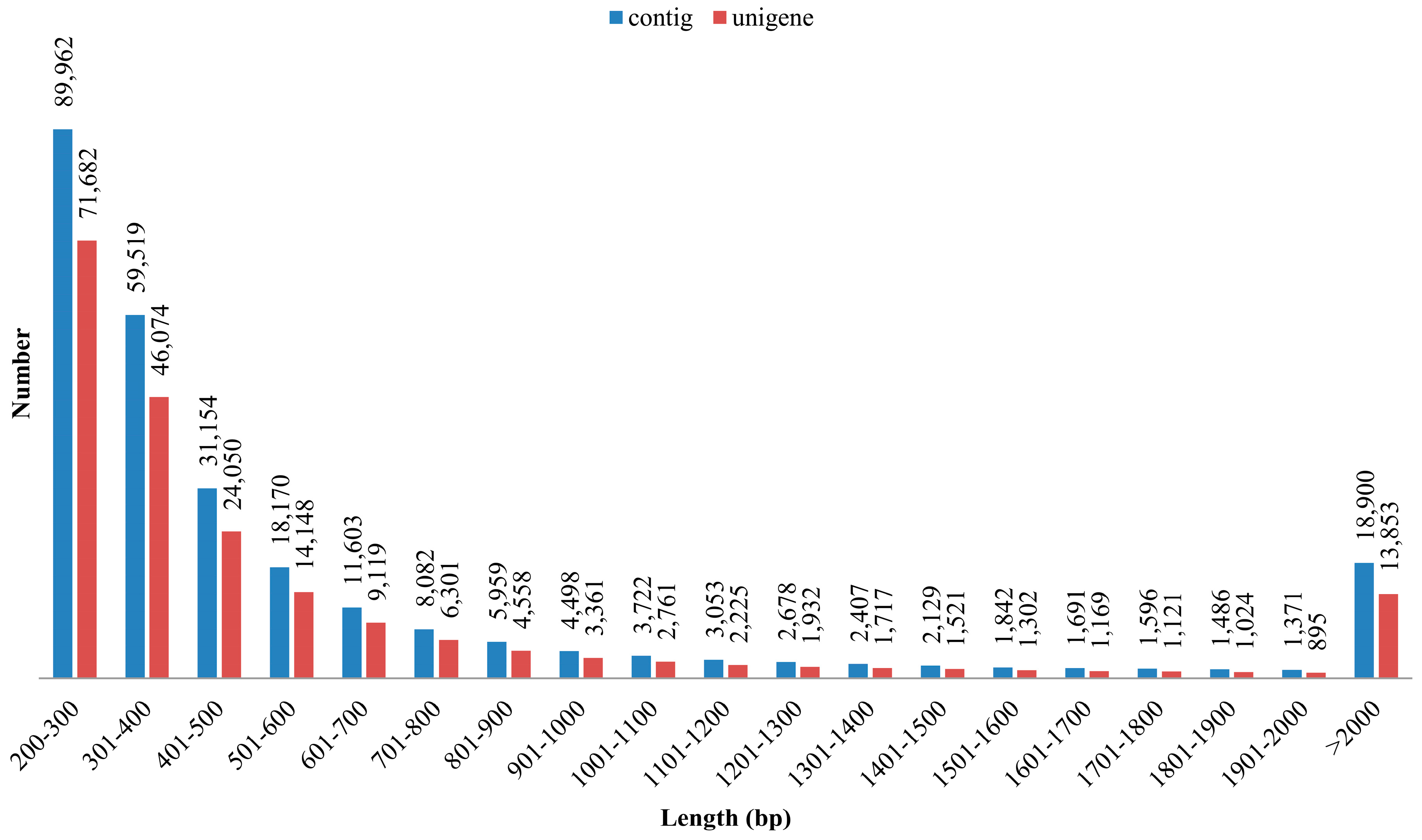
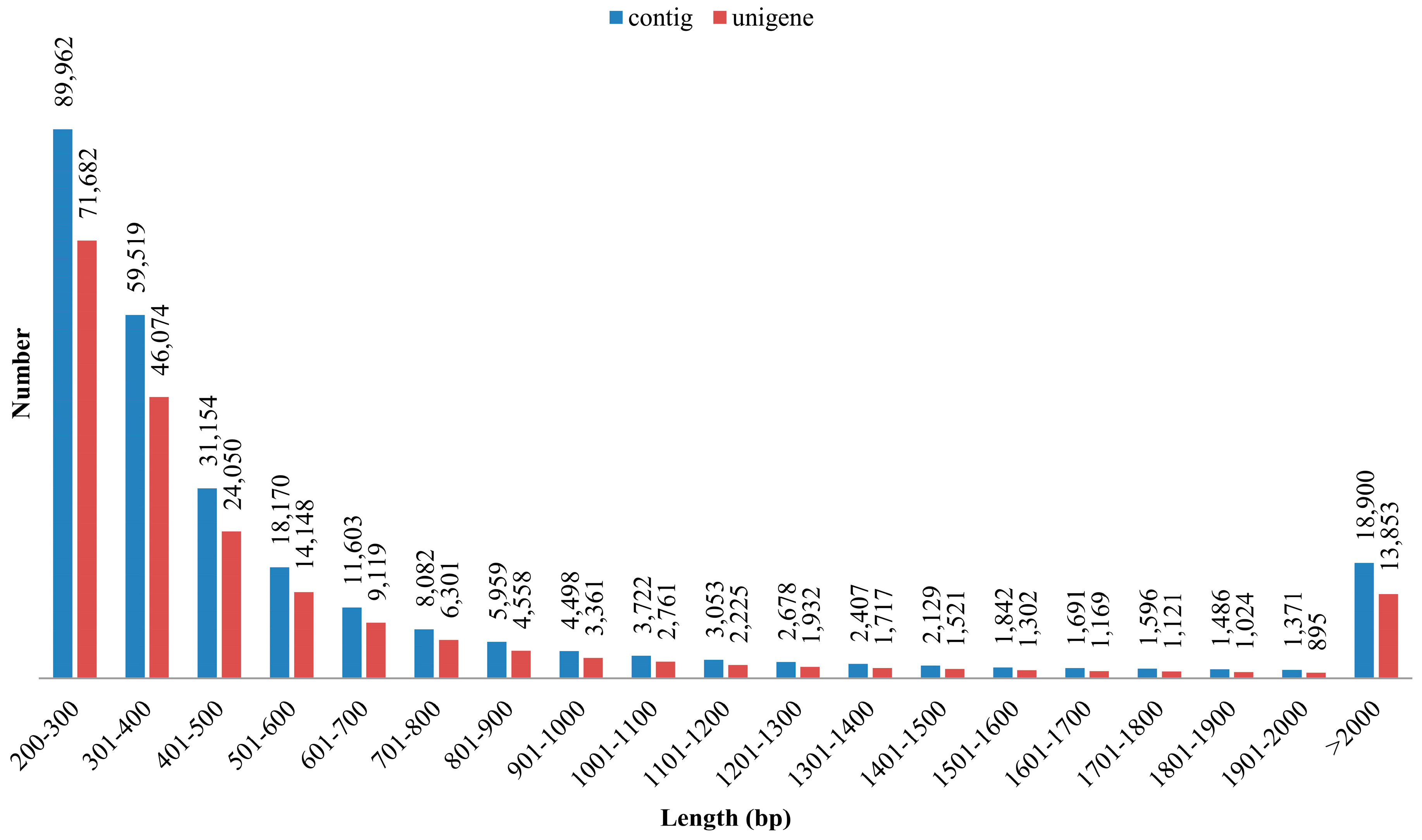
3.1. Illumina Reads and Sequence Assembly
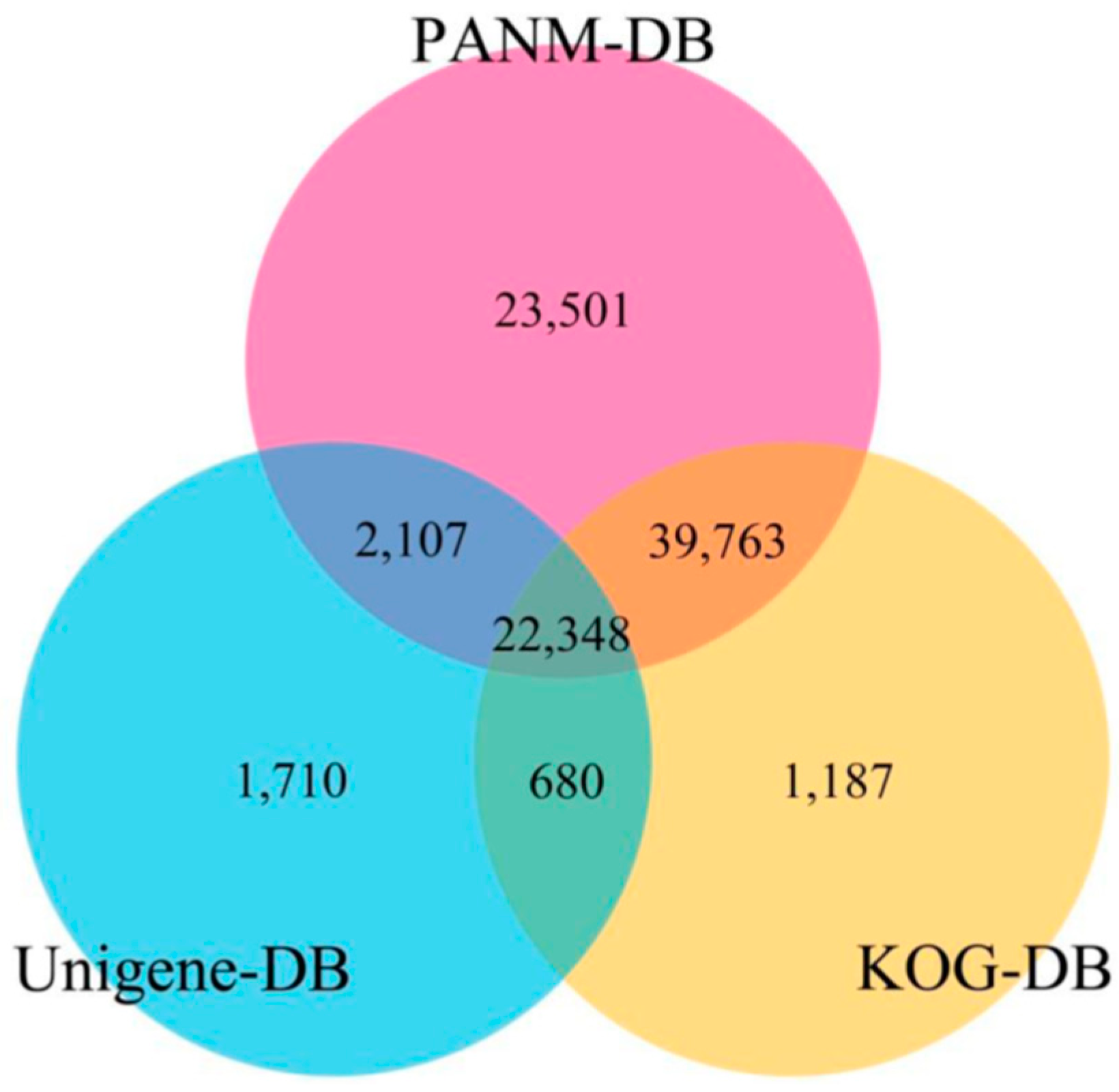
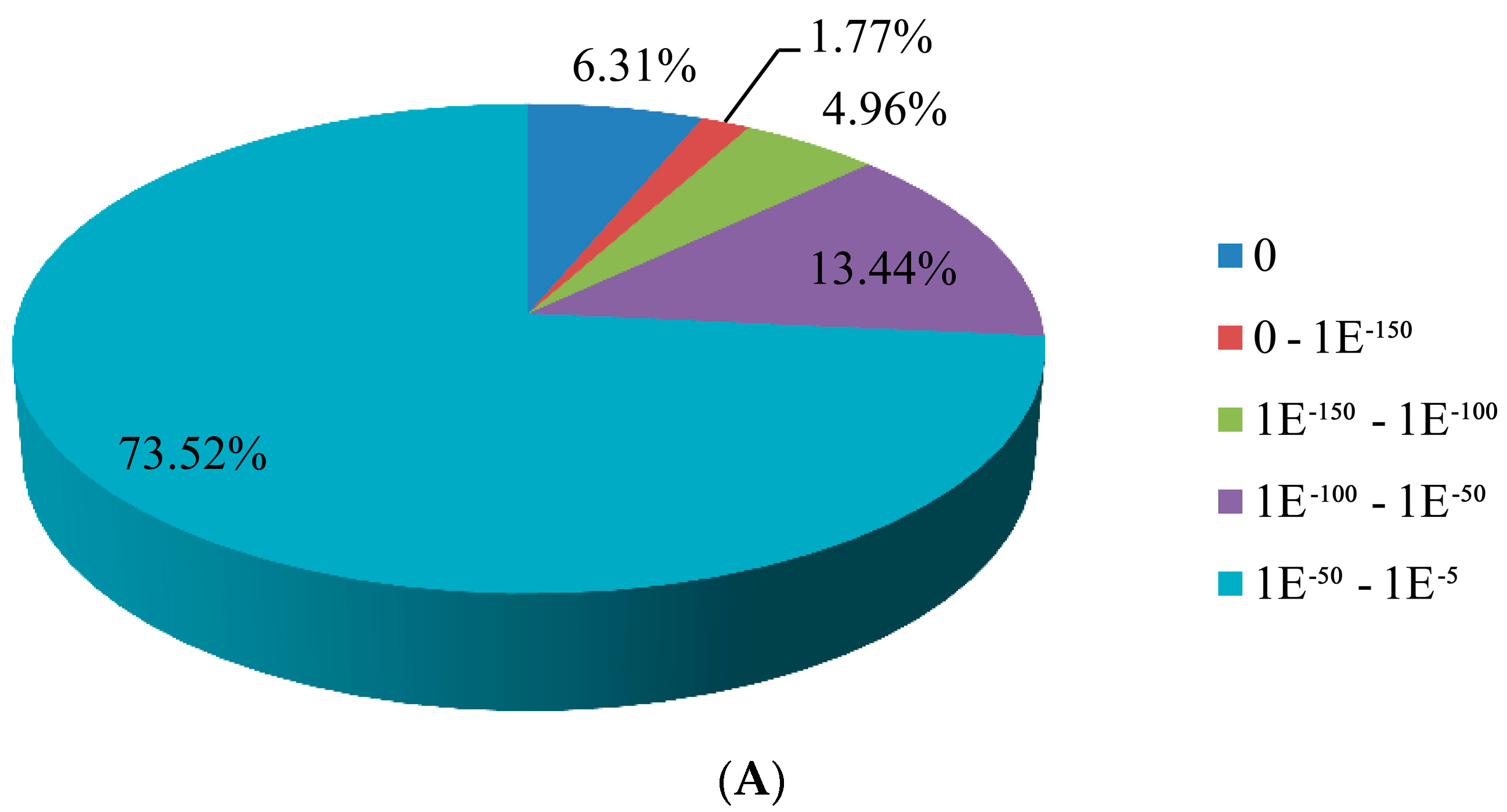
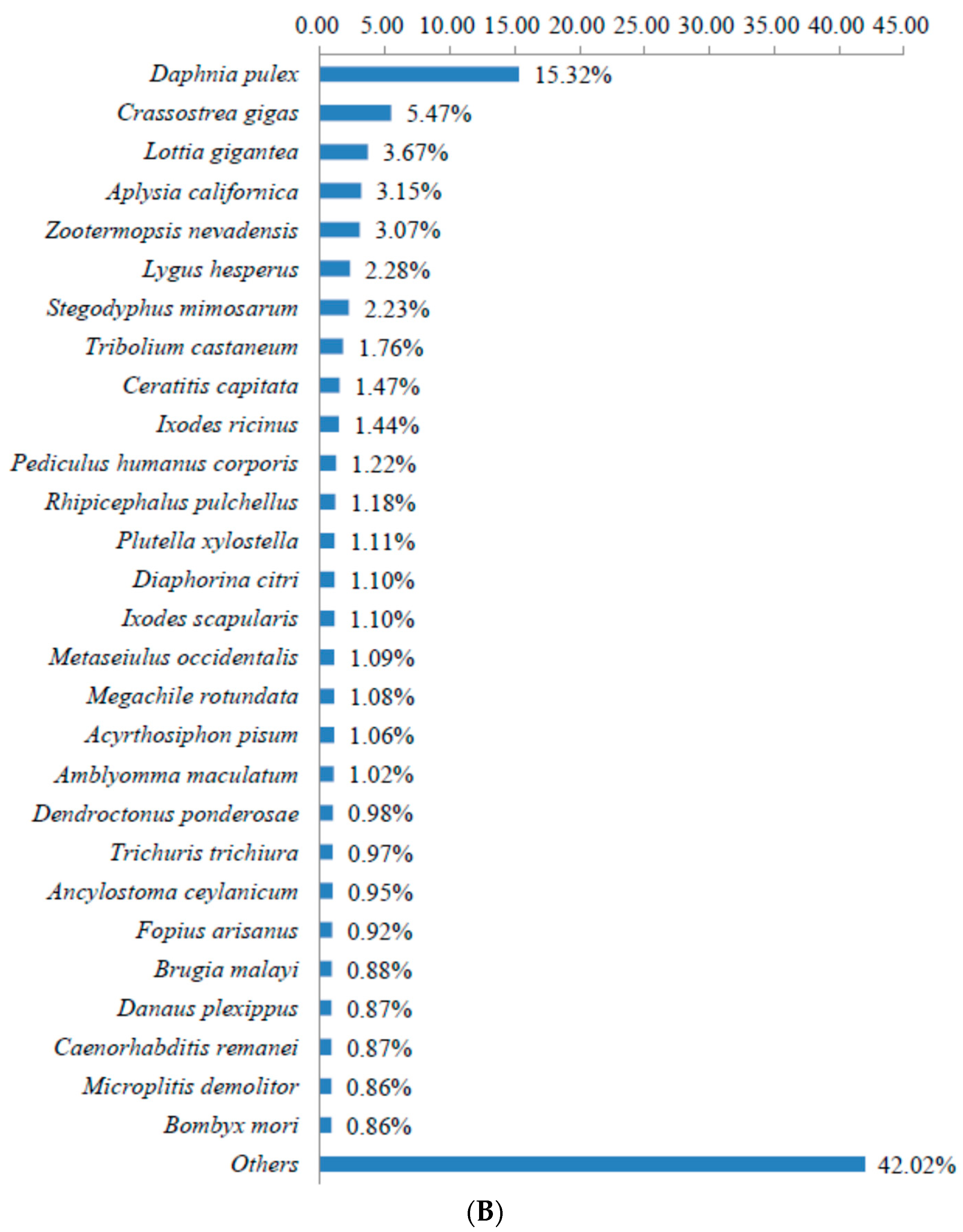
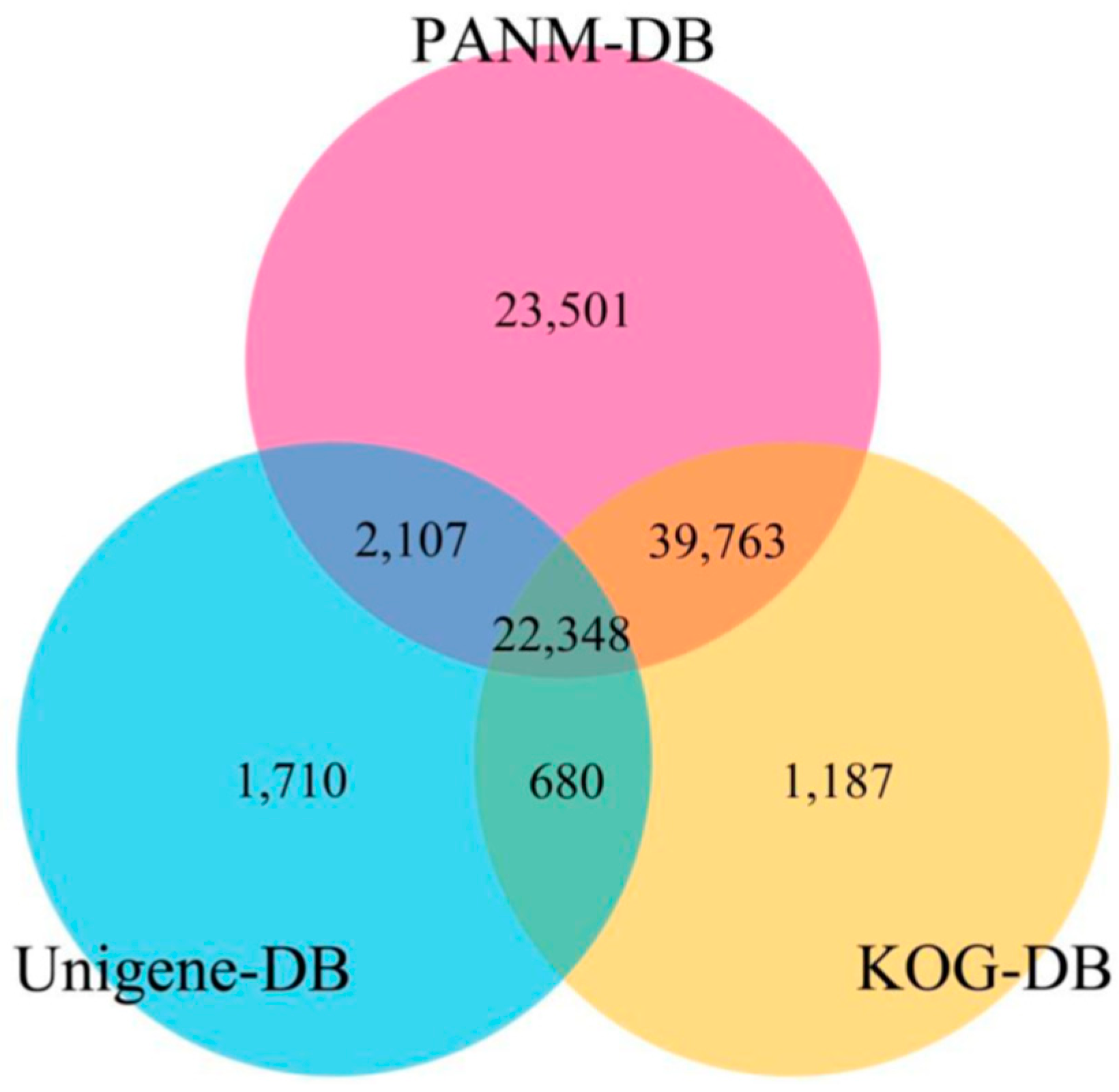
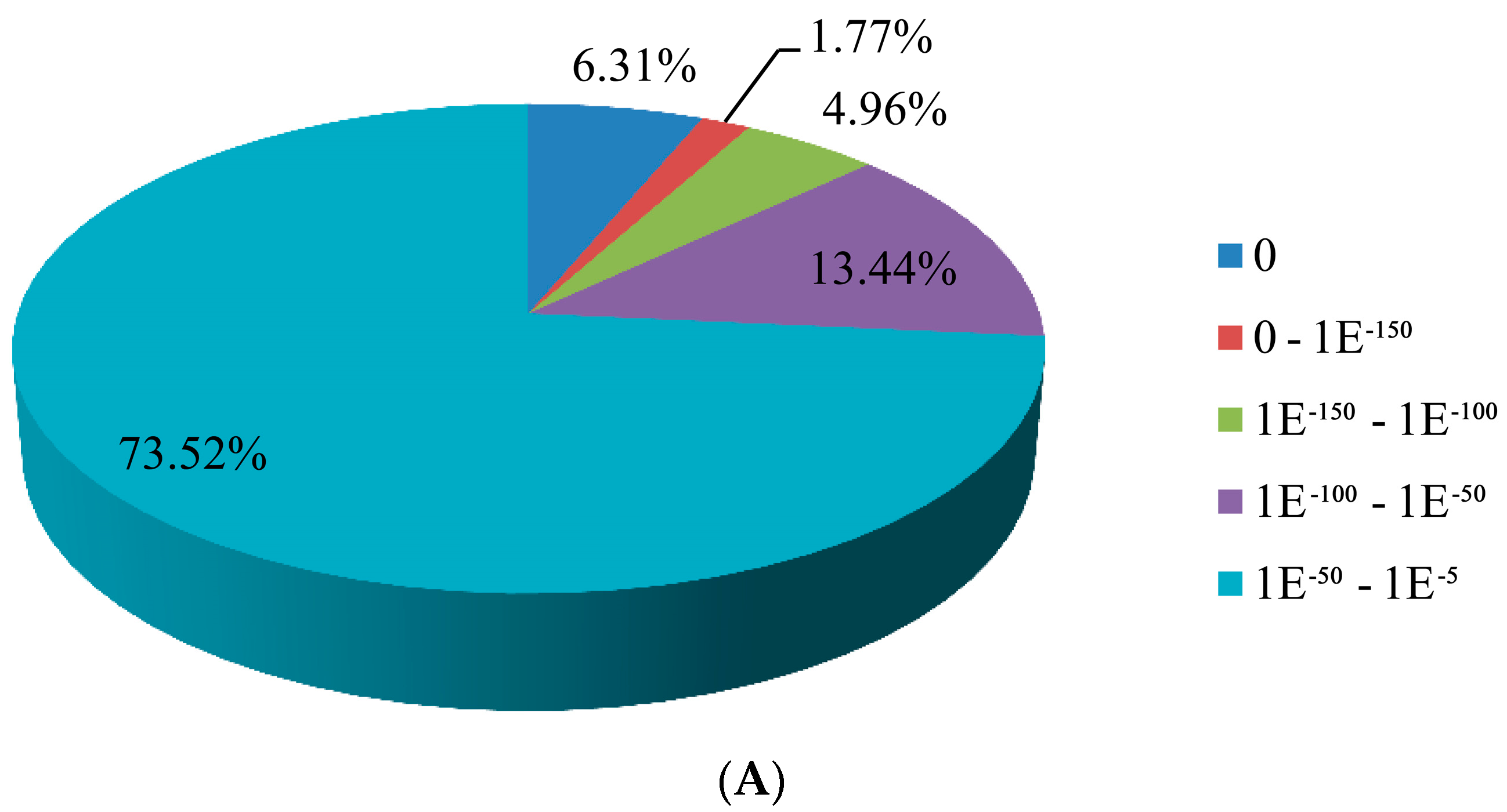
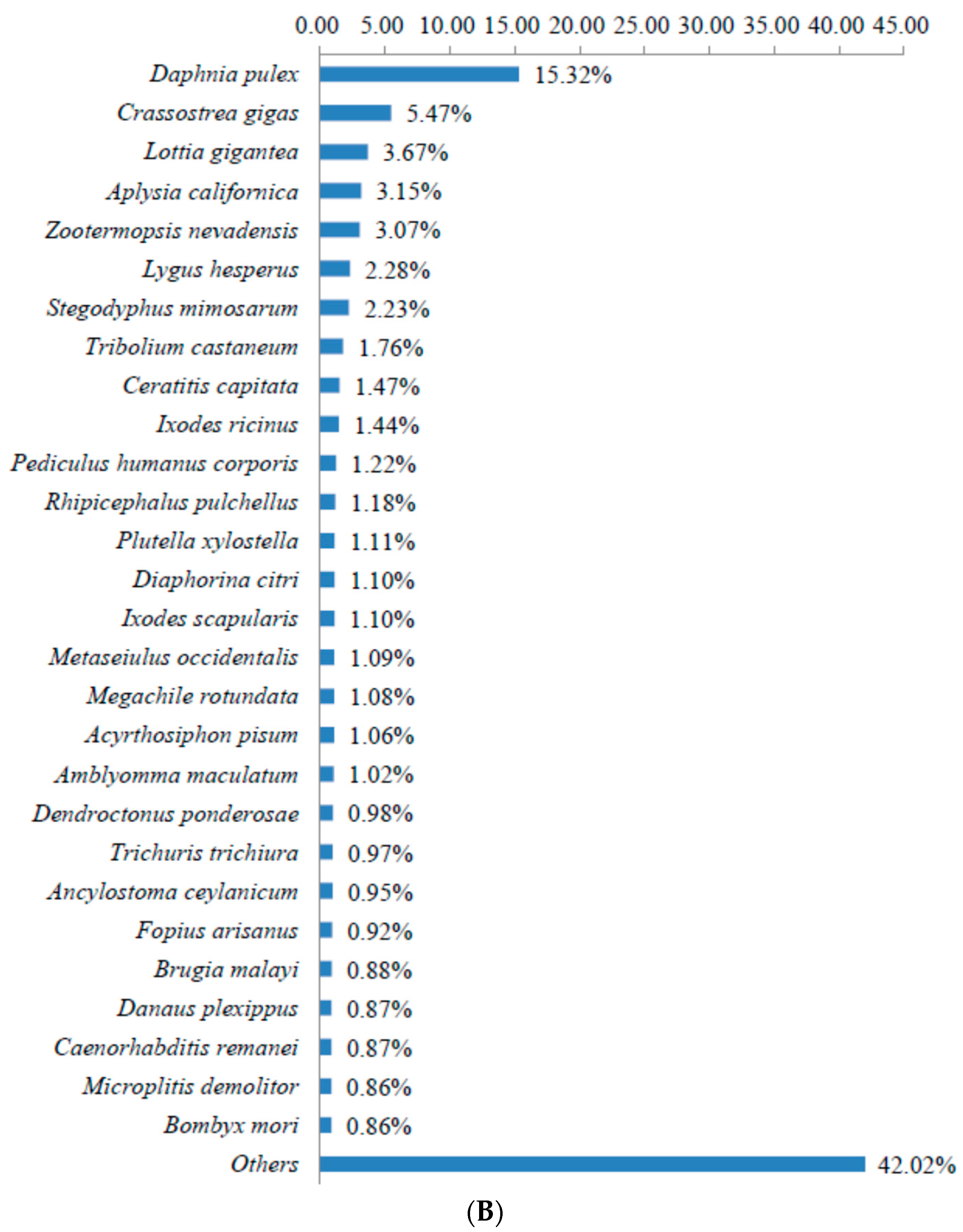
3.2. Sequence Annotation of Unigenes
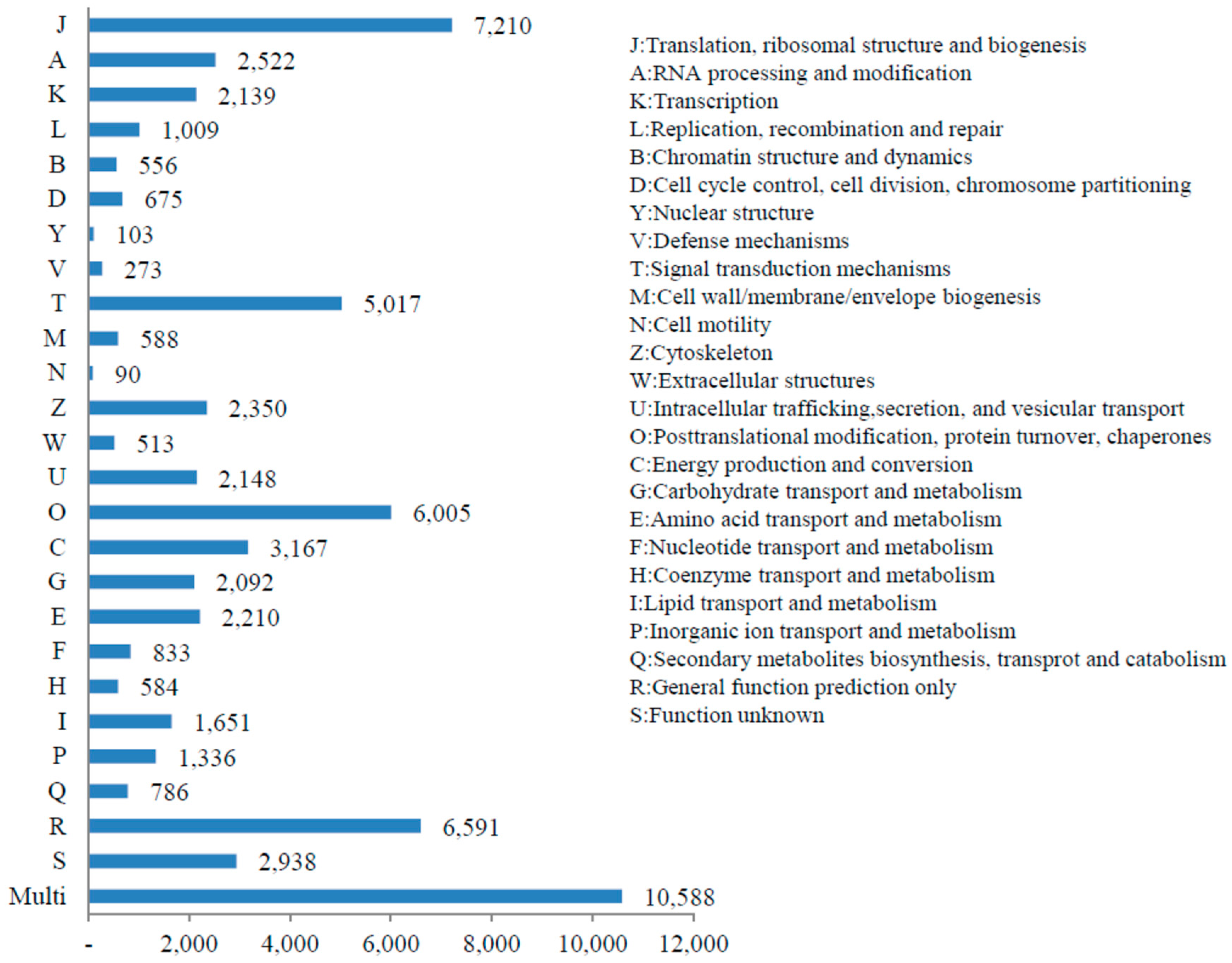
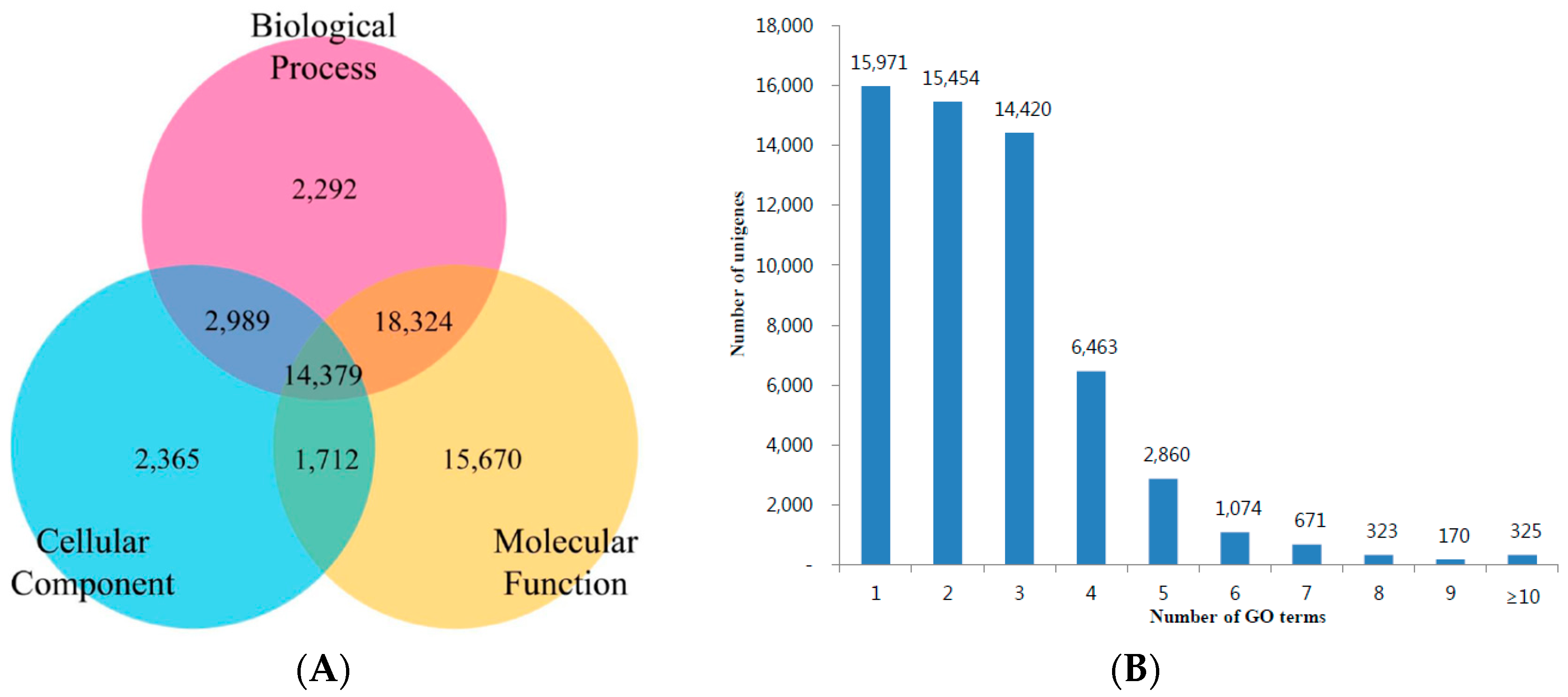
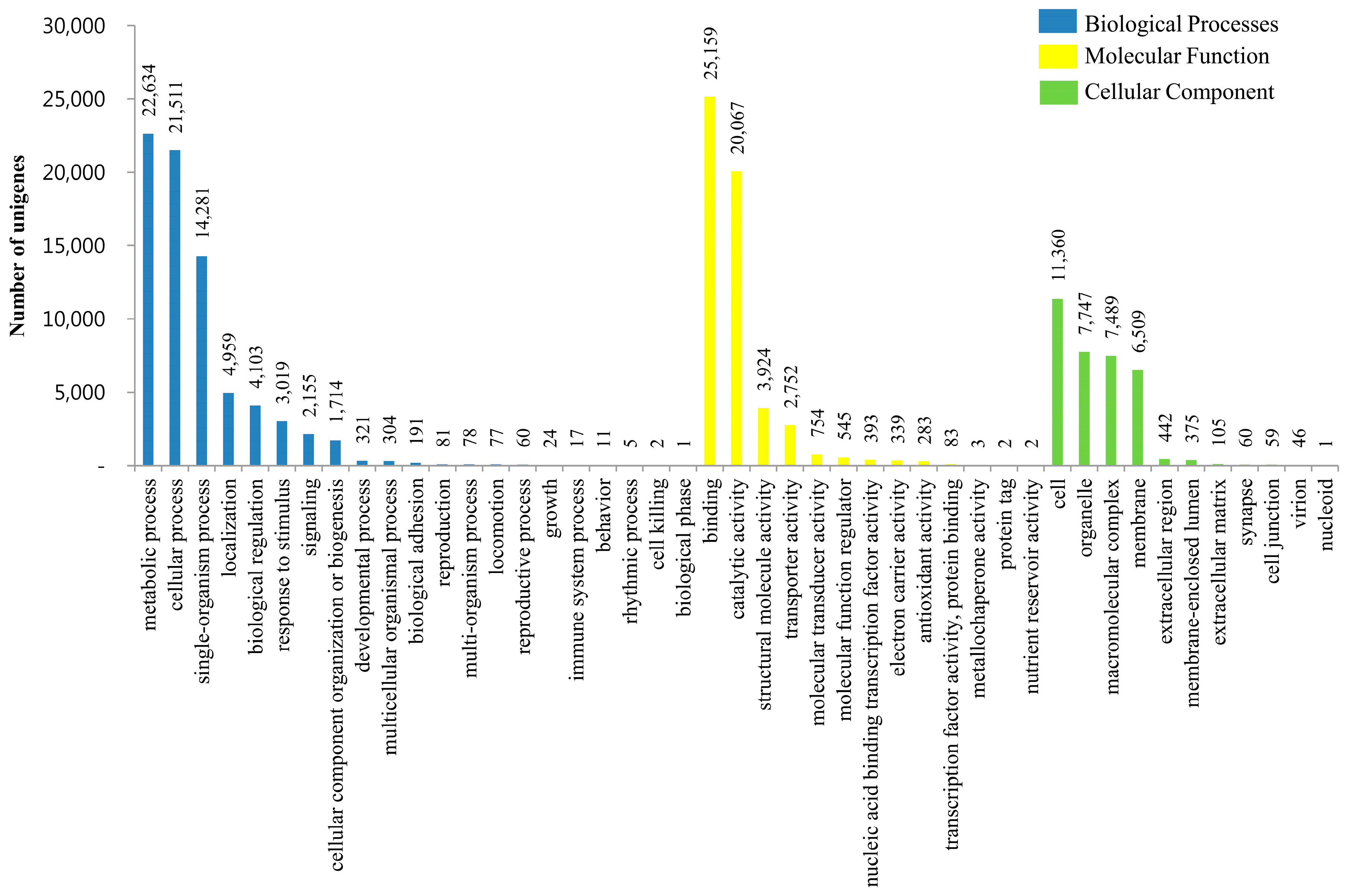
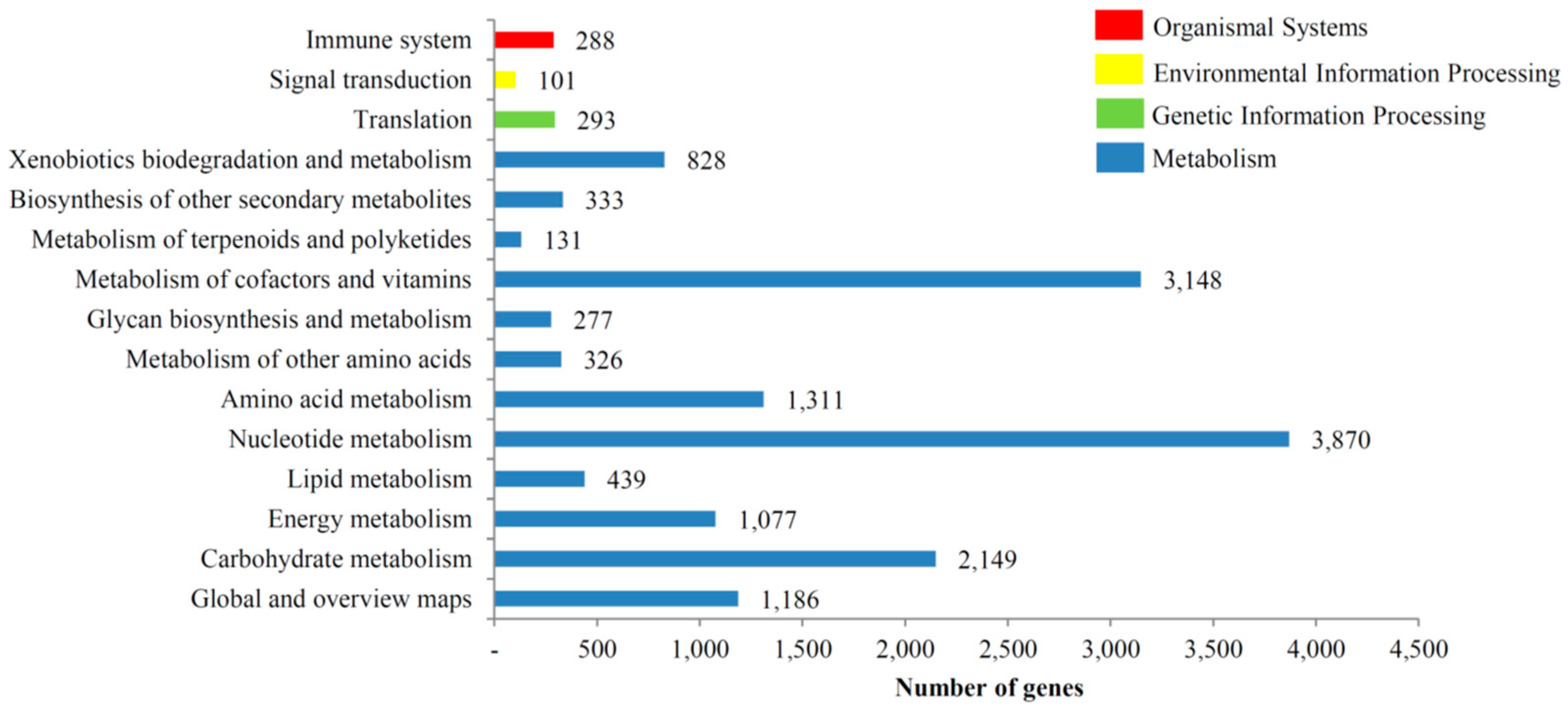
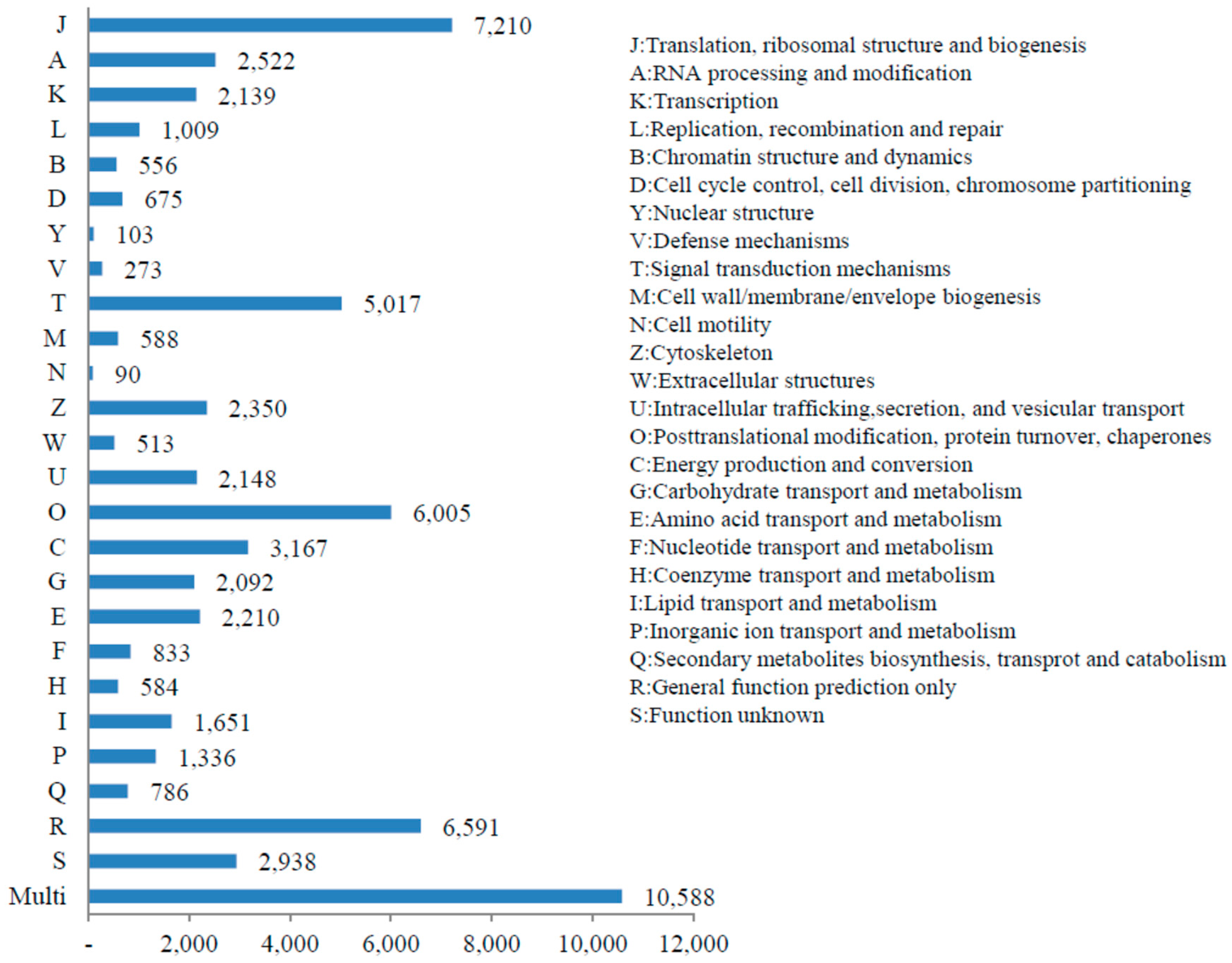
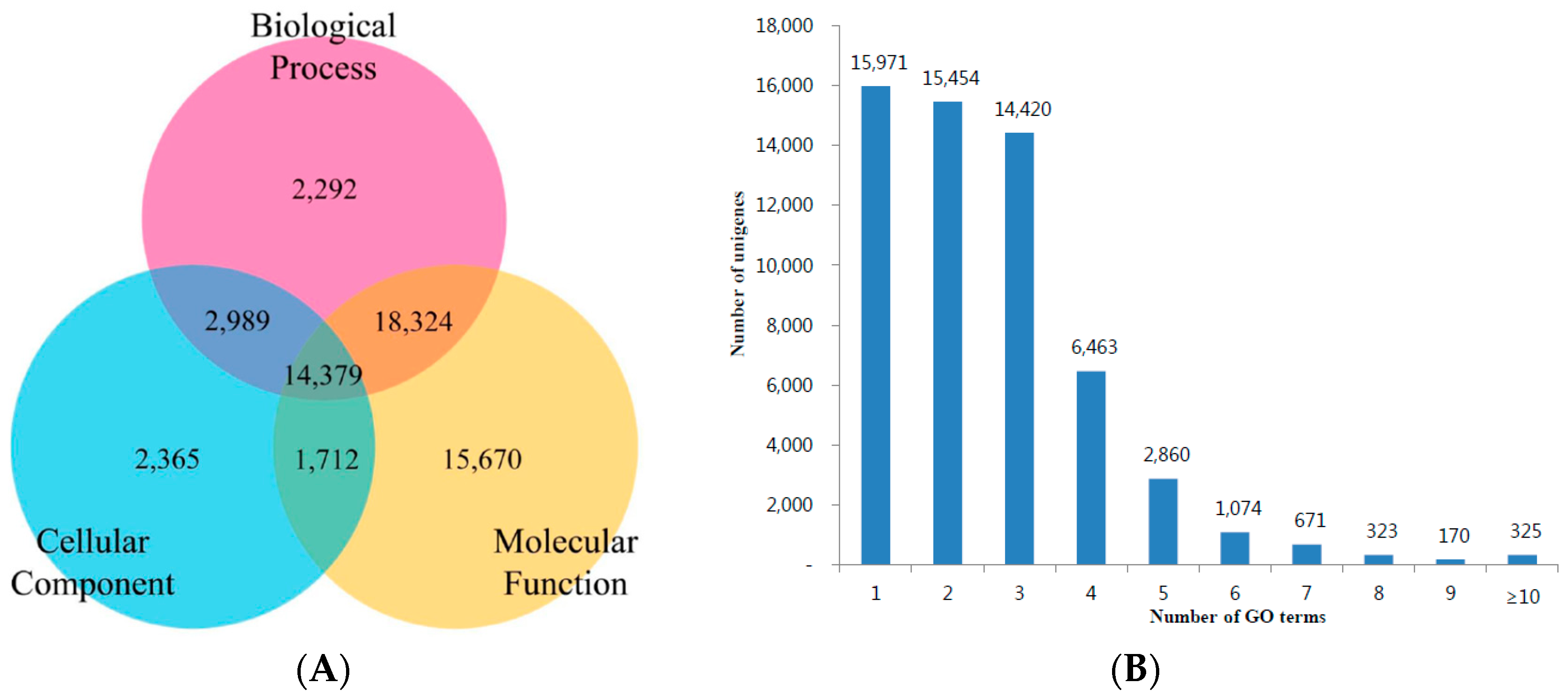
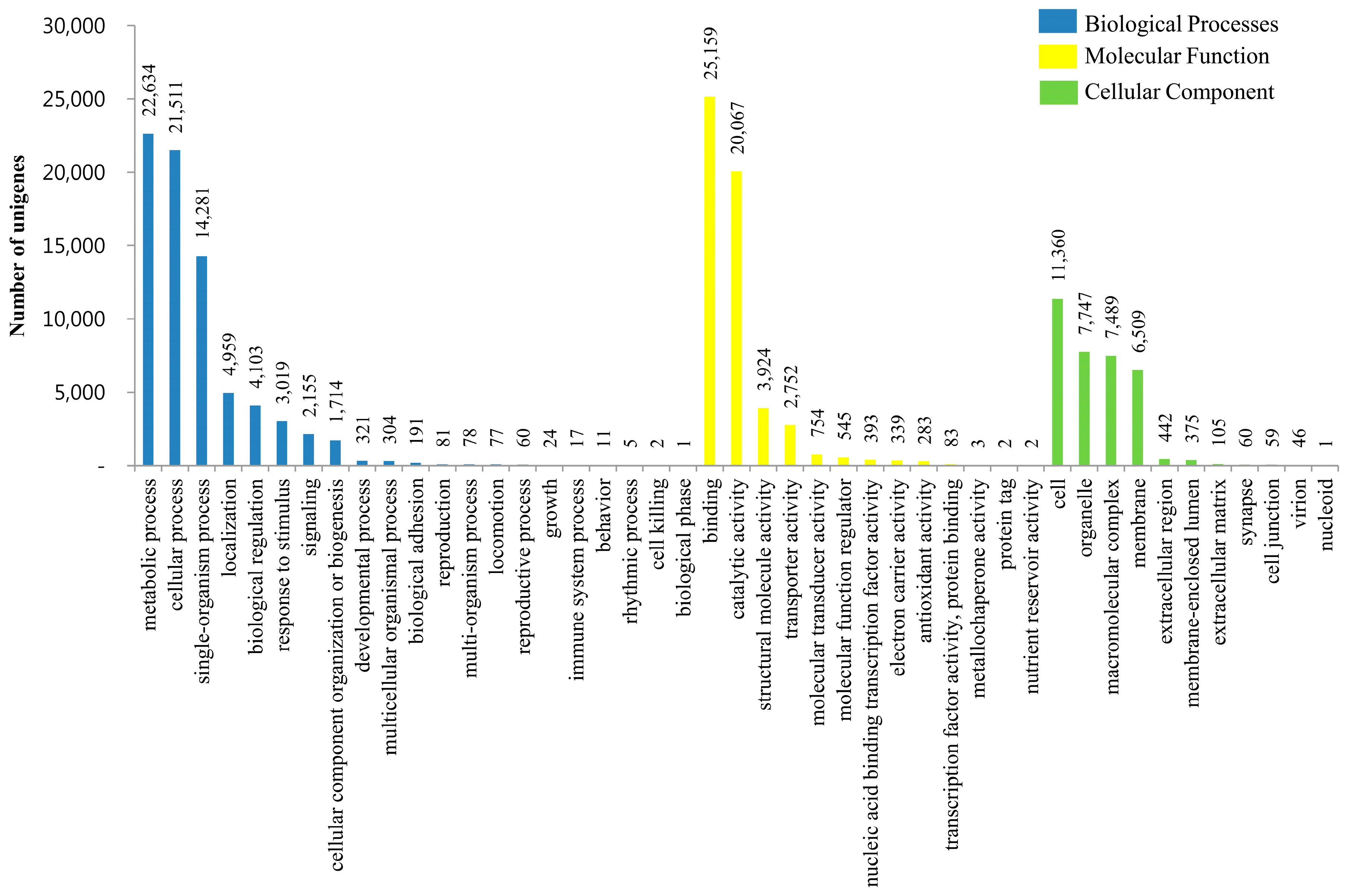
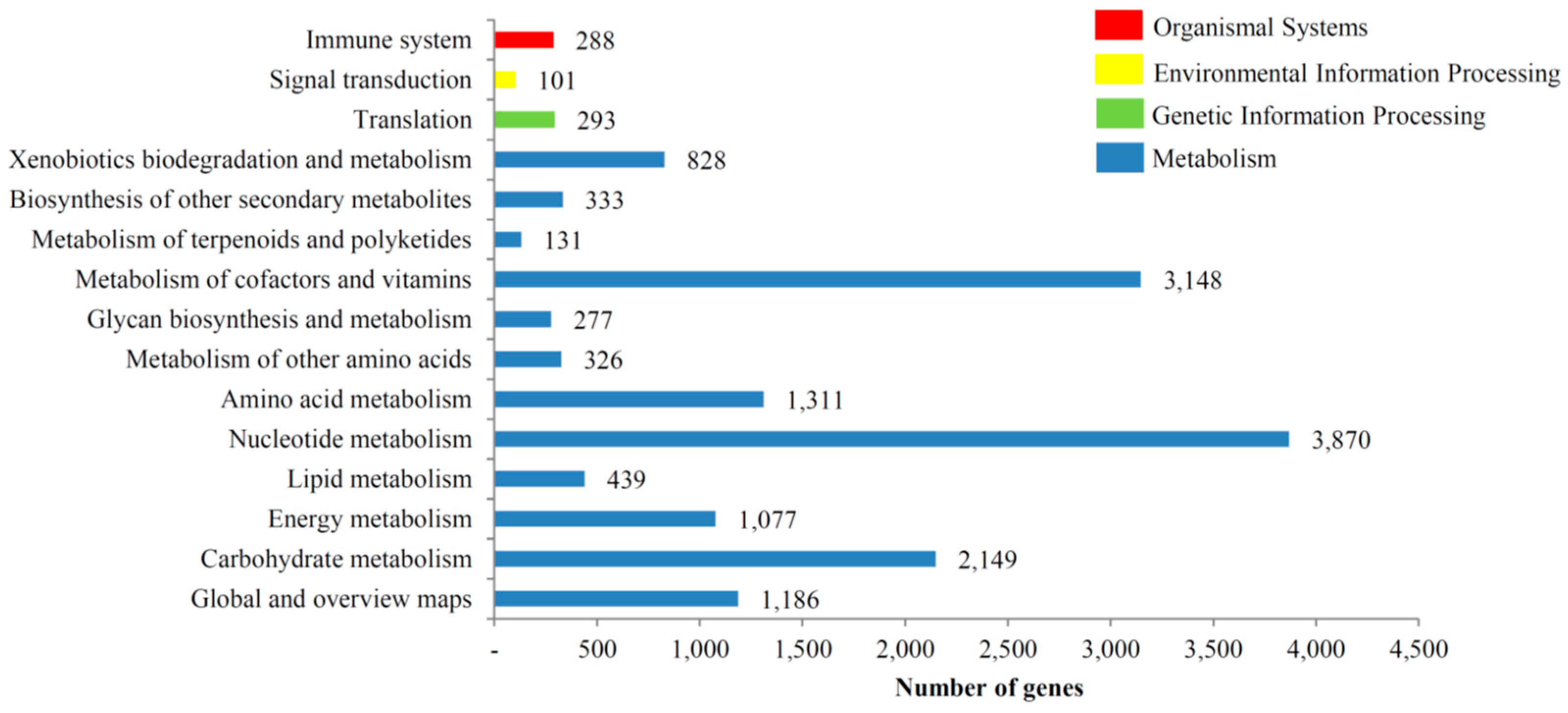
3.3. KOG, GO and KEGG Classifications
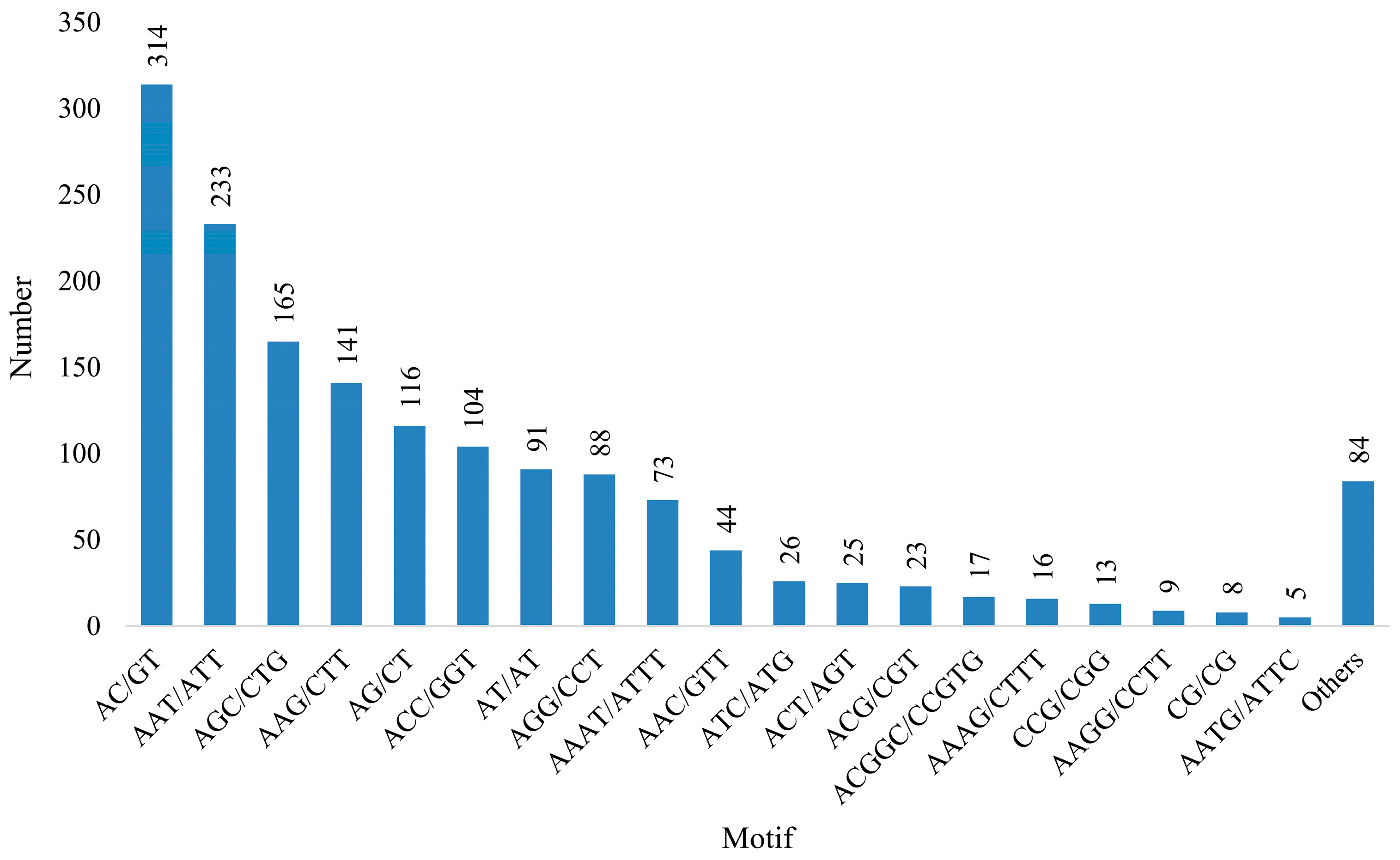
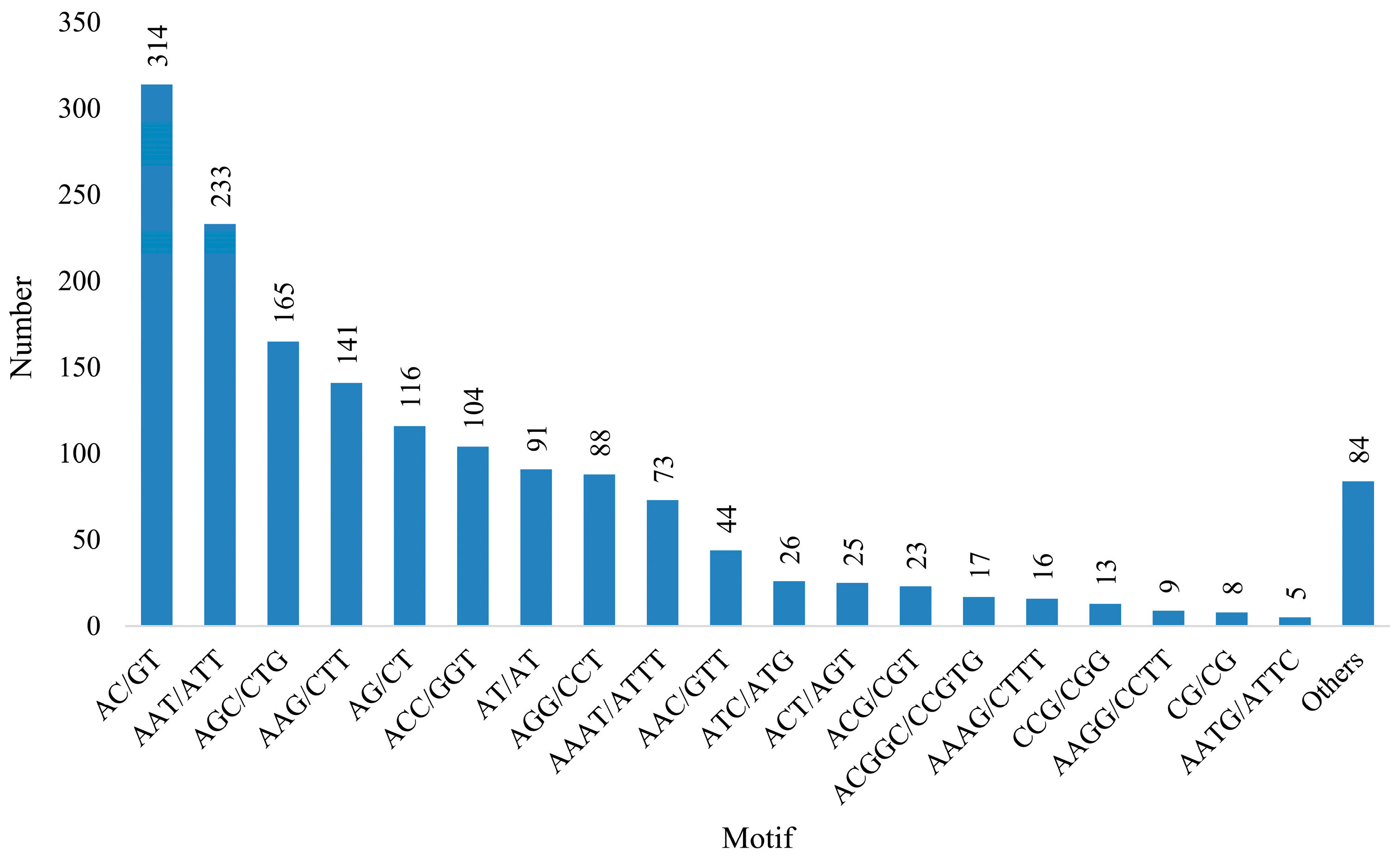
3.4. Development and SSR Locus Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Horn, R.L.; Ramaraj, T.; Devitt, N.P.; Schilkey, F.D.; Cowley, D.E. De novo assembly of a tadpole shrimp (Triops newberryi) transcriptome and preliminary differential gene expression analysis. Mol. Ecol. Resour. 2016. [Google Scholar] [CrossRef] [PubMed]
- Suno-Uchi, N.; Sasaki, F.; Chiba, S.; Kawata, M. Morphological stasis and phylogenetic relationships in Tadpole shrimps, Triops (Crustacea: Notostraca). Biol. J. Linn. Soc. 1997, 61, 439–457. [Google Scholar] [CrossRef]
- Wooten, D. Triops longicaudatus. In Zooplankton of the Great Lakes; Central Michigan University: Mount Pleasant, MI, USA, 2010. [Google Scholar]
- Fry, L.L.; Mulla, M.S.; Adams, C.W. Field introductions and establishment of the Tadpole shrimp, Triops longicaudatus (Notostraca: Triopsidae), a biological control agent of mosquitoes. Biol. Control 1994, 4, 113–124. [Google Scholar] [CrossRef]
- Becker, N.; Petric, D.; Zgomba, M.; Boase, C.; Madon, M.; Dahl, C.; Kaiser, A. Mosquitoes and Their Control; Springer-Verlag: Heidelberg, Germany, 2010. [Google Scholar]
- Yonekura, M. Weeding efficacy of tadpole shrimp (Triops spp.) in transplanted rice fields. In In Proceedings of the 7th Asian Pacific Weed Science Society Conference, Sydney, Australia, 26–30 November 1979; p. 240.
- Korn, M.; Marrone, F.; Perez-Bote, J.L.; Machado, M.; Cristo, M.; Da Fonseca, L.C.; Hundsdoerfer, A.K. Sister species within the Triops cancriformis lineage (Crustacea, Notostraca). Zool. Scr. 2006, 35, 301–322. [Google Scholar] [CrossRef]
- Macdonald, K.S.; Sallenave, R.; Cowley, D.E. Morphologic and Genetic variation in Triops (Branchiopoda: Notostraca) from ephemeral waters of the Northern Chihuahuan Desert of North America. J. Crustacean Biol. 2011, 31, 468–484. [Google Scholar] [CrossRef]
- Horn, R.L.; Cowley, D.E. Evolutionary relationships within Triops (Branchiopoda: Notostraca) using complete mitochondrial genomes. J. Crustacean Biol. 2014, 34, 795–800. [Google Scholar] [CrossRef]
- Linder, F. Contributions to the morphology and taxonomy of the Branchiopoda Notostraca, with special reference to the North American species. Proc. U. S. Natl. Mus. 1952, 102, 1–69. [Google Scholar] [CrossRef]
- Longhurst, A.R. A review of the Notostraca. Bulletin of the British Museum (Natural History). Zoology 1955, 3, 1–57. [Google Scholar]
- Akita, M. Classification of Japanese tadpole shrimps. Zool. Mag. 1976, 85, 237–247. [Google Scholar]
- Yoon, S.M.; Kim, W.; Kim, H.S. Re-description of Triops longicaudatus (LeConte, 1846) (Notostraca, Triopsidae) from Korea. Korean J. Syst. Zool. 1992, 3, 59–66. [Google Scholar]
- Sassaman, C.; Simovich, M.; Fugate, M. Reproductive isolation and genetic differentiation in North American species of Triops (Crustacea: Branchiopoda: Notostraca). Hydrobiologia 1997, 359, 125–147. [Google Scholar] [CrossRef]
- Kwon, S.J.; Jun, Y.C.; Park, J.H.; Won, D.H.; Seo, E.W.; Lee, J.E. Distribution and habitat characteristics of tadpole Shrimp. Korean J. Limnol. 2010, 43, 142–149. [Google Scholar]
- Aloufi, A.B.A.; Obuid-Allah, A.H. New records and Redescription of the notostracan Tadpole shrimp, Triops longicaudatus (LeConte, 1846) from temporary water bodies in North West region (Tabuk and Madinah) in Saudi Arabia. Int. J. Adv. Res. 2014, 2, 1222–1231. [Google Scholar]
- Kwon, S.J.; Kwon, H.J.; Jun, Y.C.; Lee, J.E.; Won, D.H. Effect of temperature on hatching rate of Triops longicaudatus (Triopsidae, Notostraca). Korean J. Limnol. 2009, 42, 32–38. [Google Scholar]
- Ryu, J.S.; Hwang, U.W. Complete mitochondrial genome of the longtail tadpole shrimp Triops longicaudatus (Crustacea, Branchiopoda, Notostraca). Mitochondrial DNA 2010, 21, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Qi, D.; Chen, L.; Zhang, H.; Zhang, X.; Qin, J.G.; Hu, S. Gene discovery from an ovary cDNA library of oriental river prawn Macrobrachium nipponense by ESTs annotation. Comp. Biochem. Physiol. Part D Genom. Proteom. 2009, 4, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, L.; Han, Y.; Ren, X.; Huang, J.; Chen, H. De novo transcriptome sequencing and analysis of Coccinella septempunctata L. in non-diapause, diapause and diapause-terminated states to identify diapause-associated genes. BMC Genom. 2015, 16, 1086. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Dong, D.; Yang, Q.; Zhu, D. Salt-responsive transcriptome profiling of Suaeda glauca via RNA Sequencing. PLoS ONE 2016, 11, e0150504. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.B.; Hwang, H.-J.; Kang, S.W.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kim, S.; et al. Transcriptome Characterization for non-model endangered Lycaenids, Protantigius superans and Spindasis takanosis, using Illumina HiSeq 2500 Sequencing. Int. J. Mol. Sci. 2015, 16, 29948–29970. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Marra, M.A. Applications of Next-Generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.T.; Malwald, F.; Schorn, C.; Bass, C.; Ott, M.C.; Nauen, R. A de novo transcriptome of European pollen beetle populations and its analysis, with special reference to insecticide action and resistance. Insect Mol. Biol. 2014, 23, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Wei, C.L.; Liu, E.T.; Ruan, Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009, 19, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Deng, Z.; Qin, B.; Liu, X.H.; Men, Z.H. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST–SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Sadamoto, H.; Takahashi, H.; Okada, T.; Kenmoku, H.; Toyota, M.; Asakawa, Y. De novo Sequencing and transcriptome analysis of the Central Nervous System of Mollusc Lymnaea stagnalis by Deep RNA Sequencing. PLoS ONE 2012, 7, e42546. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Sun, Y.; Wang, R.; Xu, T. Transcriptomic analysis of endangered Chinese salamander: Identification of immune, sex and reproduction-related genes and genetic markers. PLoS ONE 2014, 9, e87940. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software]. Available online: https://github.com/najoshi/sickle (accessed on 13 July 2016).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Ge, G.B.; Xiao, P.G.; Zhang, Y.Y.; Yang, L. The first insight into the Taxus genome via fosmid library construction and end sequencing. Mol. Genet. Genom. 2011, 285, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Patnaik, B.B.; Hwang, H.J.; Park, S.Y.; Lee, J.S.; Han, Y.S.; Lee, Y.S. PANM DB (Protosome DB) for the annotation of NGS data of mollusks. Korean J. Malacol. 2015, 31, 243–247. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadapoulous, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, E.; Xu, Z.; Li, T.; Xu, C.; Qin, J.G.; Chen, L. Comparative Transcriptome analysis in the hepatopancreas tissue of Pacific White Shrimp Litopenaeus vannamei fed different lipid sources at low salinity. PLoS ONE 2015, 10, e0144889. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Knibb, W.; Remilton, C.; Elizur, A. De-novo transcriptome analysis of the banana shrimp (Renneropenaeus merguiensis) and identification of genes associated with reproduction and development. Mar. Genom. 2015, 22, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Miranda, D.; Gallardo-Escarate, C.; Valenzuela-Munoz, V.; Farlora, R.; Gajardo, G. Sex-dependent transcriptome analysis and single nucleotide polymorphism (SNP) discovery in the brine shrimp Artemia franciscana. Mar. Genom. 2014, 18 PB, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, B.; Bickley, L.K.; Santos, E.M.; Tyler, C.R.; Stentiford, G.D.; Bateman, K.S.; van Aerle, R. De novo assembly of the Carcinus maenus transcriptome and characterization of innate immune system pathways. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ma, C.; Li, S.; Jiang, W.; Li, X.; Liu, Y.; Ma, L. Transcriptome analysis of the Mud Crab (Scylla paramamosain) by 454 deep sequencing: Assembly, Annotation and Marker Discovery. PLoS ONE 2014, 9, e102668. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, P.; Gao, B.; Wang, Y.; Wang, Z.; Chen, P.; Li, J. Transcriptome analysis of the Portunus trituberculatus: De novo assembly, growth-related gene identification and marker discovery. PLoS ONE 2014, 9, e94055. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ye, C.X.; Wang, A.L.; Xian, J.A.; Liao, S.A.; Miao, Y.T.; Zhang, S.P. Transcriptome analysis of the Pacific white shrimp Litopenaeus vannamei exposed to nitrite by RNA-seq. Fish Shellfish Immunol. 2013, 35, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Pavasovic, A.; Mather, P.B.; Prentis, P.J. Transcriptome analysis and characterization of gill-expressed carbonic anhydrase and other key osmoregulatory genes in freshwater crayfish Cherax quadricarinatus. Data Brief 2015, 5, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Ryouka, K.-M.; Wada, K.; Azuma, N.; Chiba, S. Expression profiling without genome sequence information in a non-model species, Pandalid shrimp (Pandalus latirostris), by next-generation sequencing. PLoS ONE 2011, 6, e26043. [Google Scholar]
- Zeng, V.; Villaneuva, K.E.; Ewen-Campen, B.S.; Alwes, F.; Browne, W.E.; Extavour, C.G. De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis. BMC Genom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Asselman, J.; Pfrender, M.E.; Lopez, J.A.; De Coninck, D.I.; Janssen, C.R.; Shaw, J.R.; De Schamophelaere, K.A. Conserved transcriptional responses to cyanobacterial stressors are mediated by alternate regulation of paralogous genes in Daphnia. Mol. Ecol. 2015, 24, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Hwang, H.J.; Park, S.Y.; Wang, T.H.; Park, E.B.; Lee, T.H.; Hwang, U.W.; Lee, J.S.; Park, H.S.; Han, Y.S.; et al. Mollusks sequence database: Version II. Korean J. Malacol. 2014, 30, 429–431. [Google Scholar] [CrossRef]
- Hwang, H.-J.; Patnaik, B.B.; Kang, S.W.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Patnaik, H.H.; Kim, C.; et al. RNA sequencing, de novo assembly, and functional annotation of an endangered Nymphalid butterfly, Fabriciana nerippe Felder, 1862. Entomol. Res. 2016, 2, 148–161. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Wood, V.; Dolinski, K.; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.B.; Wang, T.H.; Kang, S.W.; Hwang, H.-J.; Park, S.Y.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kim, S.; et al. Sequencing, de novo assembly, and annotation of the transcriptome of the endangered freshwater pearl bivalve, Cristaria plicata, provides novel insights into functional genes and marker discovery. PLoS ONE 2016, 11, e0148622. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Patnaik, B.B.; Kang, S.W.; Hwang, H.-J.; Chung, J.M.; Song, D.K.; Sang, M.K.; Patnaik, H.H.; Lee, J.B.; Noh, M.Y.; et al. Transcriptome analysis of the endangered neritid species Clithon retropictus: De novo Assembly, Functional Annotation, and Marker Discovery. Genes 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, E.; Li, T.; Xu, C.; Wang, X.; Lin, H.; Qin, J.G.; Chen, L. Transcriptome and pathway analysis of the Hepatopancreas in the Pacific White Shrimp Litopenaeus vannamei under Chronic Low-Salinity stress. PLoS ONE 2015, 10, e0131503. [Google Scholar] [CrossRef] [PubMed]
- Pathak, E.; Atri, N.; Mishra, R. Role of highly central residues of P-loop and it’s flanking region in preserving the archetypal conformation of Walker A motif of diverse P-loop NTPases. Bioinformation 2013, 9, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Razin, S.V.; Borunova, V.V.; Maksimenko, O.G.; Kantidze, O.L. Cys2His2 Zinc Finger Protein Family: Classification, Functions, and Major Members. Biochemistry (Moscow) 2012, 77, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xiao, S.; Ye, H.; Zhang, Z.; Lv, C.; Zheng, S.; Wang, Z.; Wang, X. Identification of immune related genes and development of SSR/SNP markers from the spleen transcriptome of Schizothorax prenanti. PLoS ONE 2016, 11, e0152572. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Yang, X.; Li, H.; Su, S.; Yi, H.; Chai, L.; Deng, X. De novo transcriptome assembly of pummelo and molecular marker development. PLoS ONE 2015, 10, e0120615. [Google Scholar] [CrossRef] [PubMed]
- La Rota, M.; Kantety, R.V.; Yu, J.K.; Sorrells, M.E. Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genom. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Annadurai, R.S.; Neethiraj, R.; Jayakumar, V.; Damodaran, A.C.; Rao, S.N.; Katta, M.A.V.S.K.; Gopinathan, S.; Sarma, S.P.; Senthilkumar, V.; Niranjan, V.; et al. De novo transcriptome assembly (NGS) of Curcuma longa L. rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLoS ONE 2013, 8, e56217. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tan, Z.; Zeng, G.; Peng, J. Comprehensive analysis of simple sequence repeats in pre-miRNAs. Mol. Biol. Evol. 2010, 27, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D.; Good, R.T.; Coleman, R.A.; Lancaster, M.L.; Weeks, A.R. Microsatellite loci and the complete mitochondrial DNA sequence characterized through next generation sequencing and de novo assembly for the critically endangered orange-bellied parrot, Neophema chrysogaster. Mol. Biol. Rep. 2013, 40, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Luo, H.; Du, H.; Wang, D.Q.; Wei, Q.W. Isolation and characterization of twenty-six microsatellite loci for the tetraploid fish Dabry’s sturgeon (Acipenser dabryanus). Conserv. Genet. Res. 2013, 5, 409–412. [Google Scholar] [CrossRef]
- Andriantahina, F.; Liu, X.; Huang, H. Genetic map construction and quantitative trait locus (QTL) detection of growth-related traits in Litopenaeus vannamei for selective breeding applications. PLoS ONE 2013, 8, e75206. [Google Scholar] [CrossRef]
- Shen, D.; Bo, W.; Xu, F.; Wu, R. Genetic diversity and population structure of the Tibetan poplar (Populus szechuanica var. tibetica) along an altitude gradient. BMC Genet. 2014, 15. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number of Raw Reads | |
| Number of sequences | 323,319,608 |
| Number of bases | 40,738,270,608 |
| Total number of clean reads | |
| Number of sequences | 318,610,596 |
| Number of bases | 39,745,513,470 |
| Mean length of contig (bp) | 124.7 |
| N50 length of contig (bp) | 126 |
| GC % of contig | 48.39 |
| High-quality reads (%) | 98.54 (sequences), 97.56 (bases) |
| Contig information | |
| Total number of contig | 269,822 |
| Number of bases | 192,327,026 |
| Mean length of contig (bp) | 712.8 |
| N50 length of contig (bp) | 1148 |
| GC % of contig | 46.82 |
| Largest contig (bp) | 40,450 |
| No. of large contigs (≥500 bp) | 89,407 |
| Unigene information | |
| Total number of unigenes | 208,813 |
| Number of bases | 146,173,633 |
| Mean length of unigene (bp) | 700.0 |
| N50 length of unigene (bp) | 1089 |
| GC % of unigene | 46.97 |
| Length ranges (bp) | 224–40,450 |
| Databases | All | ≤300 bp | 300–1000 bp | ≥1000 bp |
|---|---|---|---|---|
| PANM-DB | 87,719 | 20,029 | 43,958 | 23,732 |
| UNIGENE | 26,845 | 6231 | 12,885 | 7729 |
| KOG | 63,978 | 12,955 | 30,892 | 20,131 |
| GO | 57,731 | 12,915 | 28,153 | 16,663 |
| KEGG | 7247 | 1735 | 3400 | 2112 |
| ALL | 95,105 | 22,935 | 48,081 | 24,089 |
| SSR parameters | Number |
|---|---|
| Total number of sequences examined | 29,547 |
| Total size of examined sequences (bp) | 75,658,821 |
| Total number of identified SSRs | 1595 |
| Di-nucleotide | 529 |
| Tri-nucleotide | 862 |
| Tetra-nucleotide | 144 |
| Penta-nucleotide | 33 |
| Hexa-nucleotide | 27 |
| Number of SSR containing sequences | 1432 |
| Number of sequences containing more than 1 SSR | 140 |
| Number of SSRs present in compound formation | 74 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seong, J.; Kang, S.W.; Patnaik, B.B.; Park, S.Y.; Hwang, H.J.; Chung, J.M.; Song, D.K.; Noh, M.Y.; Park, S.-H.; Jeon, G.J.; et al. Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery. Genes 2016, 7, 114. https://doi.org/10.3390/genes7120114
Seong J, Kang SW, Patnaik BB, Park SY, Hwang HJ, Chung JM, Song DK, Noh MY, Park S-H, Jeon GJ, et al. Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery. Genes. 2016; 7(12):114. https://doi.org/10.3390/genes7120114
Chicago/Turabian StyleSeong, Jiyeon, Se Won Kang, Bharat Bhusan Patnaik, So Young Park, Hee Ju Hwang, Jong Min Chung, Dae Kwon Song, Mi Young Noh, Seung-Hwan Park, Gwang Joo Jeon, and et al. 2016. "Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery" Genes 7, no. 12: 114. https://doi.org/10.3390/genes7120114
APA StyleSeong, J., Kang, S. W., Patnaik, B. B., Park, S. Y., Hwang, H. J., Chung, J. M., Song, D. K., Noh, M. Y., Park, S.-H., Jeon, G. J., Kong, H. S., Kim, S., Hwang, U. W., Park, H. S., Han, Y. S., & Lee, Y. S. (2016). Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery. Genes, 7(12), 114. https://doi.org/10.3390/genes7120114