Somatic Mosaicism in the Human Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Somatic Mosaicism
1.1. Early Studies of Mosaicism
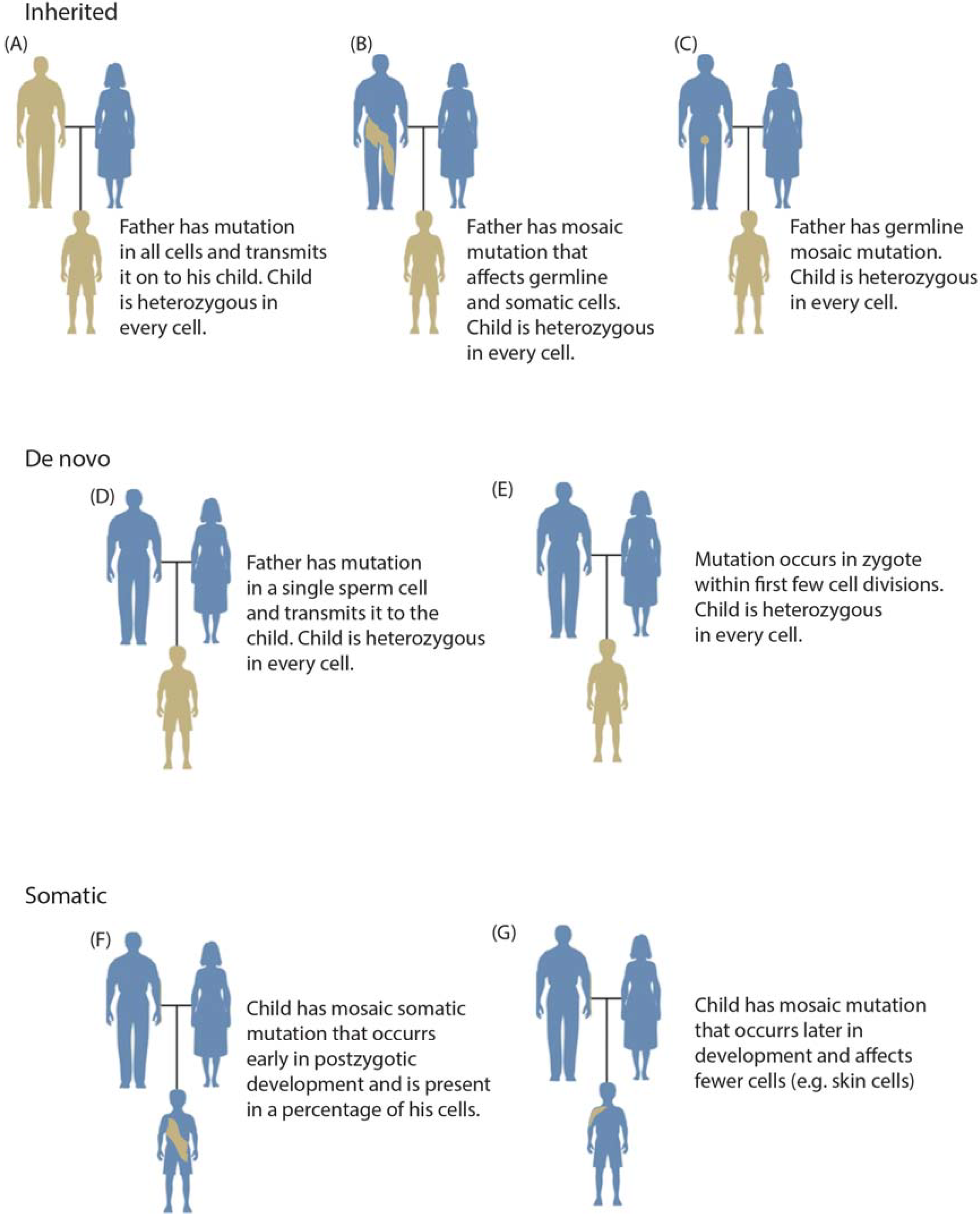
1.2. Categories of Somatic Variation
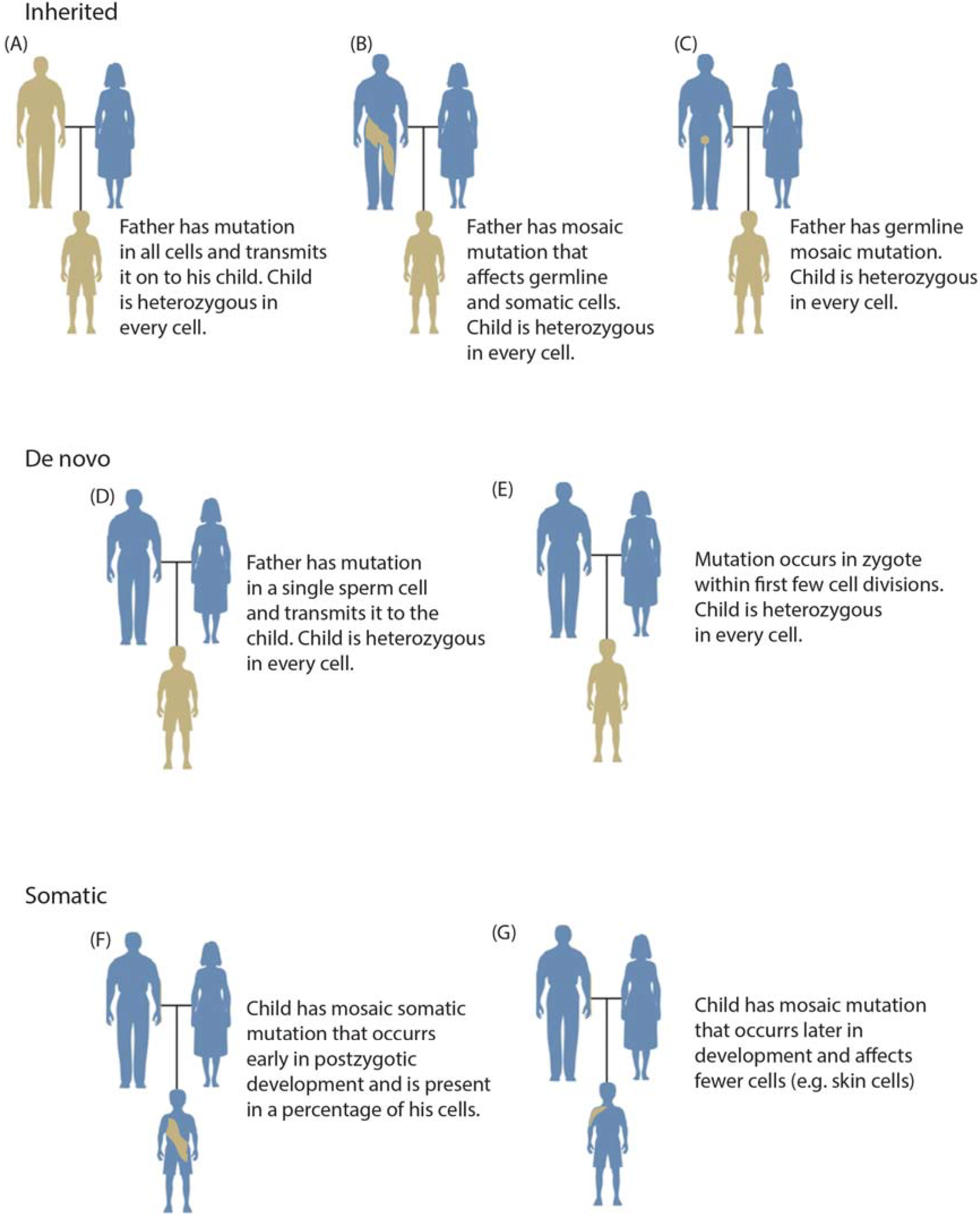
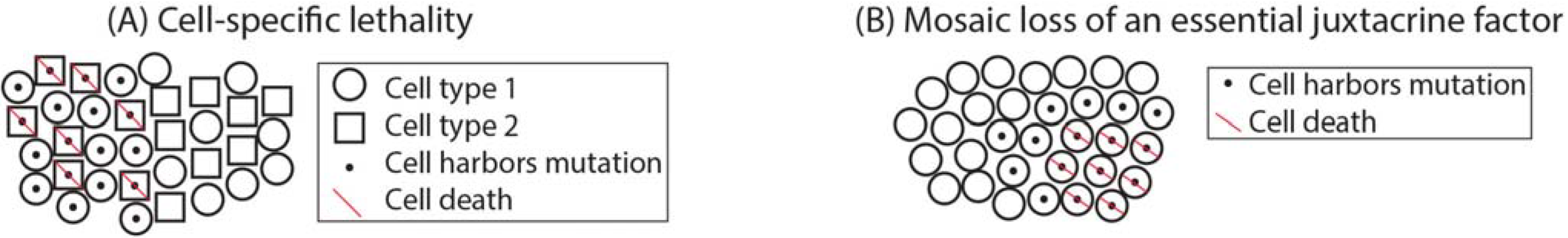
1.3. Mosaicism during Development
1.4. Mosaicism across the Body
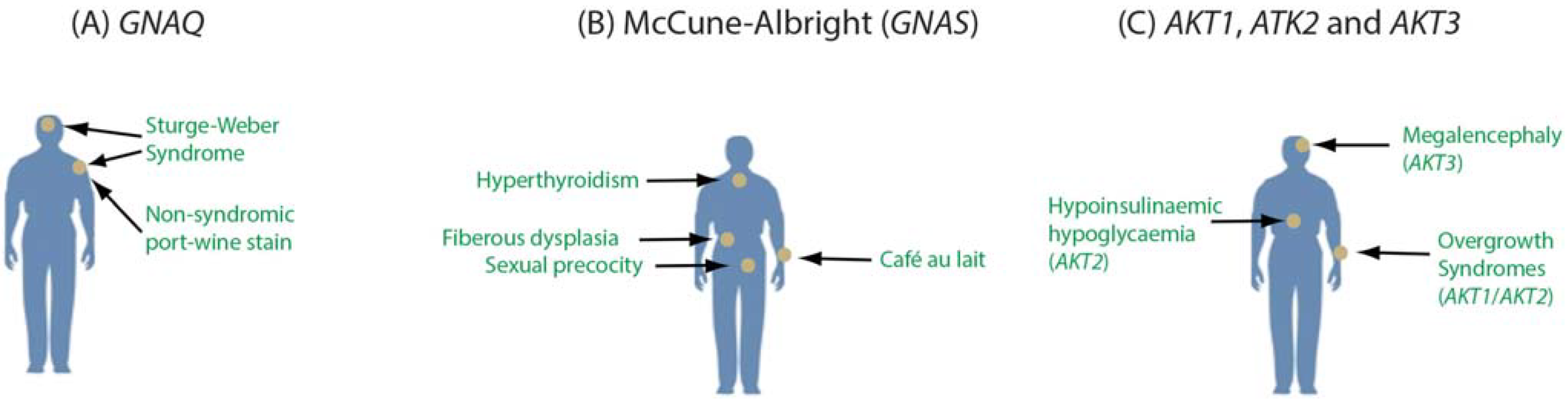
2. Detection of Somatic Mosaicism
2.1. Technical Considerations
2.2. Cytogenetics
2.3. Genome-Wide Arrays
2.4. Second-Generation Sequencing
3. Somatic Mosaicism in Disease
3.1. Cancer and Aging
3.2. Neurodegenerative Disease
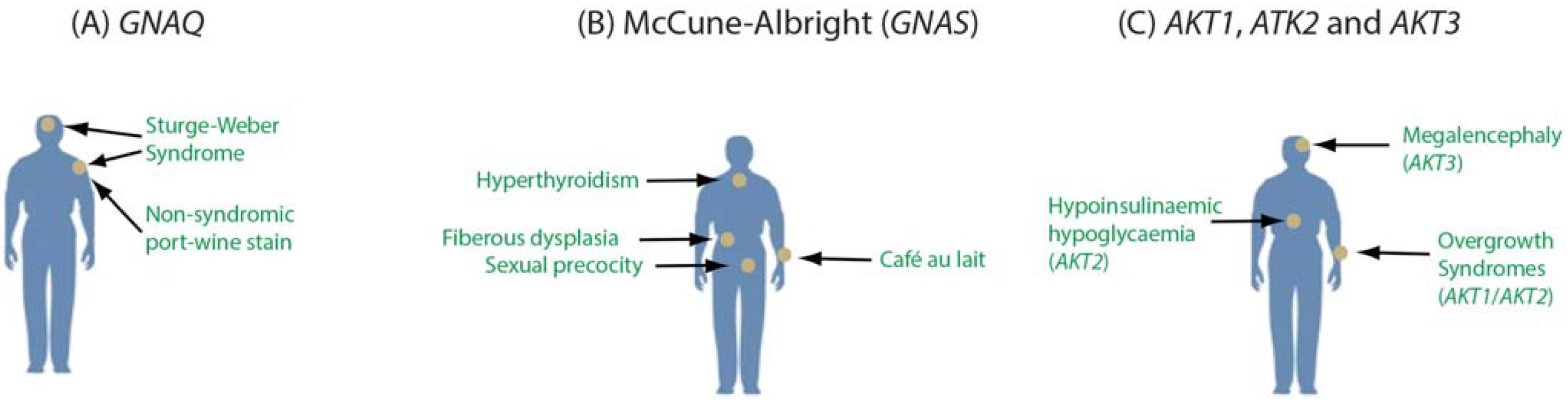
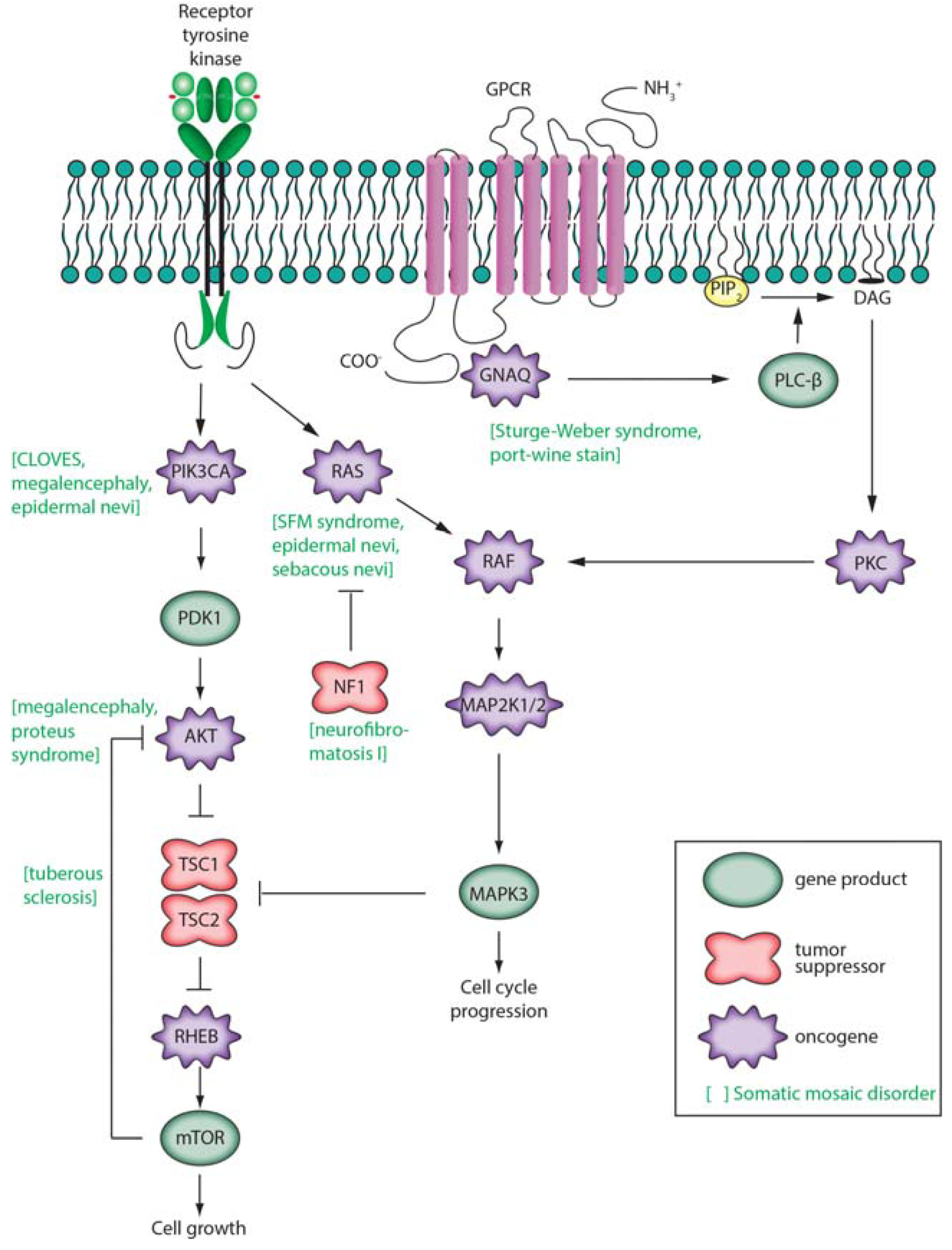
3.3. Monogenic Disease
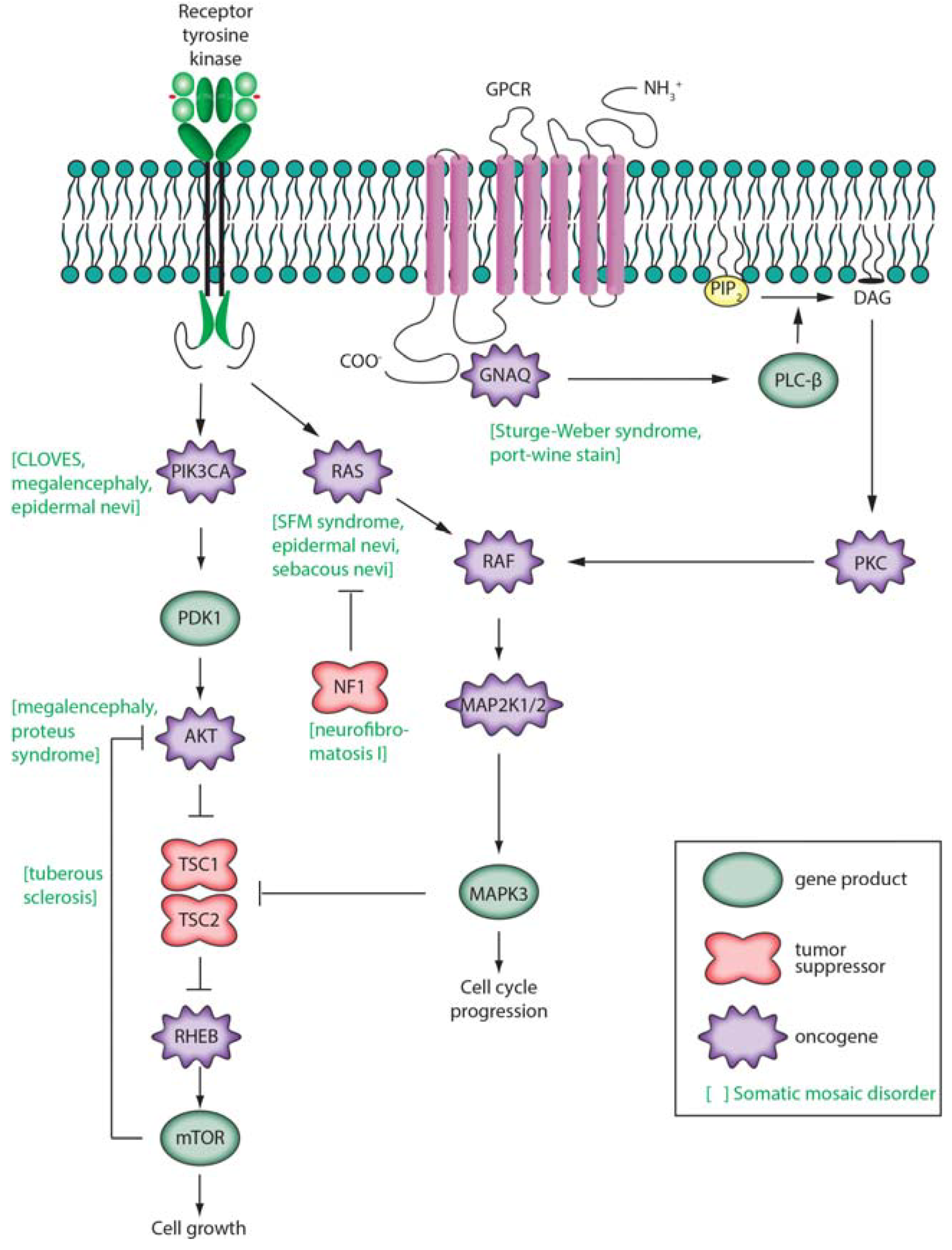
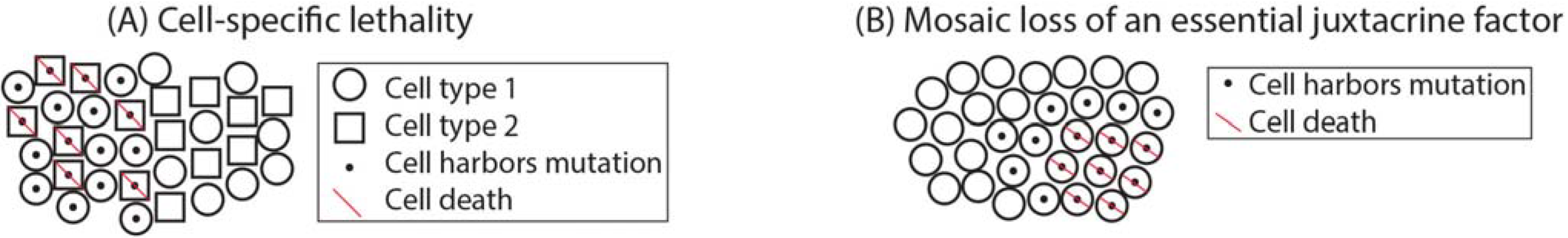
3.4. Complex Disease
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Edwards, J.H. Familiarity, recessivity and germline mosaicism. Ann. Hum. Genet. 1989, 53, 33–47. [Google Scholar] [CrossRef]
- Hartl, D.L. Recurrence risks for germinal mosaics. Am. J. Hum. Genet. 1971, 23, 124–134. [Google Scholar] [PubMed]
- Poduri, A.; Evrony, G.D.; Xuyu, C.; Walsh, C.A. Somatic mutation, genomic variation, and neurological disease. Science 2013, 341. [Google Scholar] [CrossRef]
- Campbell, I.M.; Yuan, B.; Robberecht, C.; Pfundt, R.; Szafranski, P.; McEntagart, M.E.; Nagamani, S.C.; Erez, A.; Bartnik, M.; Wisniowiecka-Kowalnik, B.; et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 2014, 95, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Van der Maarel, S.M.; Deidda, G.; Lemmers, R.J.L.F.; van Overveld, P.G.M.; van der Wielen, M.; Hewitt, J.E.; Sandkuijl, L.; Bakker, B.; van Ommen, G.-J.B.; Padberg, G.W.; et al. De novo facioscapulohumeral muscular dystrophy: Frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am. J. Hum. Genet. 2000, 66, 26–35. [Google Scholar]
- Boveri, T. The Origin of Malignant Tumors; The Williams and Wilkins Company: Baltimore, MD, USA, 1929. [Google Scholar]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. Chromosome organization and genic expression. Cold Spring Harb. Symp. Quant. Biol. 1951, 16, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M. The Clonal Selection Theory of Acquired Immunity; Vanderbilt University Press: Nashville, TN, USA, 1959. [Google Scholar]
- Brack, C.; Hirama, M.; Lenhard-Schuller, R.; Tonegawa, S. A complete immunoglobulin gene is created by somatic recombination. Cell 1978, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Krangel, M.S. Mechanics of t cell receptor gene rearrangement. Curr. Opin. Immunol. 2009, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Frumkin, D.; Wasserstrom, A.; Kaplan, S.; Feige, U.; Shapiro, E. Genomic variability within an organism exposes its cell lineage tree. PLOS Comput. Biol. 2005, 1, e50. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P. Somatic gene mutation and human disease other than cancer: An update. Mutat. Res. 2010, 705, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P. Recent advances in the study of somatic mosaicism and diseases other than cancer. Curr. Opin. Genet. Dev. 2014, 26, 73–78. [Google Scholar] [CrossRef]
- Erickson, R.P. Somatic gene mutation and human disese other than cancer. Mutat. Res. 2003, 543, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, R. In vivo reversion to normal of inherited mutations in humans. J. Med. Genet. 2003, 40, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F.; Castellanos Nuijts, M.; van Essen, A.J. Natural repair mechanisms in correcting pathogenic mutations in inherited skin disorders. Clin. Exp. Dermatol. 2003, 28, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Lai-Cheong, J.E.; McGrath, J.A.; Uitto, J. Revertant mosaicism in skin: Natural gene therapy. Trends Mol. Med. 2011, 17, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F. Revertant mosaicism in human genetic disorders. Am. J. Med. Genet. 1999, 85, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Happle, R. Lethal genes surviving by mosaicism: A possible explanation for sporadic birth defects involving the skin. J. Am. Acad. Dermatol. 1987, 16, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Carvalho, C.M.; Hastings, P.J.; Lupski, J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012, 22, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.P. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 2000, 22, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kotzot, D. Complex and segmental uniparental disomy updated. J. Med. Genet. 2008, 45, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T. Cytogenetic contribution to uniparental disomy (UPD). Mol. Cytogenet. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H., Jr. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [PubMed]
- van den Hurk, J.A.; Meij, I.C.; Seleme, M.C.; Kano, H.; Nikopoulos, K.; Hoefsloot, L.H.; Sistermans, E.A.; de Wijs, I.J.; Mukhopadhyay, A.; Plomp, A.S.; et al. L1 retrotransposition can occur early in human embryonic development. Hum. Mol. Genet. 2007, 16, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J.; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Mirkin, S.M. The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 2013, 23, 280–288. [Google Scholar] [CrossRef] [PubMed]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010, 11, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Hellenbroich, Y.; Schwinger, E.; Zühlke, C.H. Limited somatic mosaicism for friedreich’s ataxia GAA triplet repeat expansions identified by small pool PCR in blood leukocytes. Acta Neurol. Scand. 2001, 103, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Hashida, H.; Goto, J.; Suzuki, T.; Jeong, S.-Y.; Masuda, N.; Ooie, T.; Tachiiri, Y.; Tsuchiya, H.; Kanazawa, I. Single cell analysis of cag repeat in brains of dentatorubral-pallidoluysian atrophy (DRPLA). J. Neurol. Sci. 2001, 190, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kahlem, P.; Djian, P. The expanded CAG repeat associated with juvenile Huntington disease shows a common origin of most or all neurons and glia in human cerebrum. Neurosci. Lett. 2000, 286, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Møllersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLOS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.-i.; Kondoh, K.; Komure, Y.; Komure, O.; Kuno, S.; Kawai, J.; Hazama, F.; Sano, A. Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (drpla). Hum. Mol. Genet. 1995, 4, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Montermini, L.; Kish, S.J.; Jiralerspong, S.; Lamarche, J.B.; Pandolfo, M. Somatic mosaicism for friedreich’s ataxia GAA triplet repeat expansions in the central nervous system. Neurology 1997, 49, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.M. Making sense of eukaryotic DNA replication origins. Science 2001, 294, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.M. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat. Rev. Genet. 2010, 11, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Van Echten-Arends, J.; Mastenbroek, S.; Sikkema-Raddatz, B.; Korevaar, J.C.; Heineman, M.J.; van der Veen, F.; Repping, S. Chromosomal mosaicism in human preimplantation embryos: A systematic review. Hum. Reprod. Update 2011, 17, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Amat, F.; Lemon, W.; Mossing, D.P.; McDole, K.; Wan, Y.; Branson, K.; Myers, E.W.; Keller, P.J. Fast, accurate reconstruction of cell lineages from large-scale fluorescence microscopy data. Nat. Methods 2014. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Bolognia, J.L.; Orlow, S.J.; Glick, S.A. Lines of blaschko. J. Am. Acad. Dermatol. 1994, 31, 157–190. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.; Tang, H.T.; Gallione, B.A.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.T.; Singer, F.R.; Eugster, E. Mccune-albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J. Rare Dis. 2012, 7 (Suppl 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Bastepe, M.; Juppner, H. Gnas locus and pseudohypoparathyroidism. Horm. Res. 2005, 63, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Evrony, G.D.; Cai, X.; Elhosary, P.C.; Beroukhim, R.; Lehtinen, M.K.; Hills, L.B.; Heinzen, E.L.; Hill, A.; Hill, R.S.; et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012, 74, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lindhurst, M.J.; Sapp, J.C.; Teer, J.K.; Johnston, J.J.; Finn, E.M.; Peters, K.; Turner, J.; Cannons, J.L.; Bick, D.; Blakemore, L.; et al. A mosaic activating mutation in AKT1 associated with the proteus syndrome. N. Engl. J. Med. 2011, 365, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Hussain, K.; Challis, B.; Rocha, N.; Payne, F.; Minic, M.; Thompson, A.; Daly, A.; Scott, C.; Harris, J.; Smillie, B.J.; et al. An activating mutation of AKT2 and human hypoglycemia. Science 2011, 334, 474. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.-J.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [PubMed]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Aghili, L.; Foo, J.; DeGregori, J.; De, S. Patterns of somatically acquired amplifications and deletions in apparently normal tissues of ovarian cancer patients. Cell Rep. 2014, 7, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Lambert, M.; Teshima, I.; Ray, P.N.; Richer, C.-L.; Dallaire, L. Monozygotic twins with 45, X/46, XY mosaicism discordant for phenotypic sex. Am. J. Med. Genet. 1998, 75, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Boelter, W.D.; Sparkes, R.S.; Lin, M.S.; Battersby, K. Monozygotic twins of discordant sex both with 45,X/46,X,idic(Y) mosaicism. Am. J. Med. Genet. 1991, 41, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, L.; Foster, R.; Shen, Y.; Parry, D.M.; McMaster, M.L.; O’Leary, M.C.; Gusella, J.F. Monozygotic twins discordant for neurofibromatosis 1. Am. J. Med. Genet. A 2010, 152A, 601–606. [Google Scholar] [CrossRef]
- Zeng, S.; Patil, S.R.; Yankowitz, J. Prenatal detection of mosaic trisomy 1q due to an unbalanced translocation in one fetus of a twin pregnancy following in vitro fertilization: A postzygotic error. Am. J. Med. Genet. A 2003, 120A, 464–469. [Google Scholar] [CrossRef]
- Helderman-van den Enden, A.T.J.M.; Maaswinkel-Mooij, P.D.; Hoogendoorn, E.; Willemsen, R.; Maat-Kievit, J.A.; Losekoot, M.; Oostra, B.A. Monozygotic twin brothers with the fragile X syndrome: Different CGG repeats and different mental capacities. J. Med. Genet. 1999, 36, 253–257. [Google Scholar] [PubMed]
- Piotrowski, A.; Bruder, C.E.; Andersson, R.; Diaz de Stahl, T.; Menzel, U.; Sandgren, J.; Poplawski, A.; von Tell, D.; Crasto, C.; Bogdan, A.; et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008, 29, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- O’Huallachain, M.; Karczewski, K.J.; Weissman, S.M.; Urban, A.E.; Snyder, M.P. Extensive genetic variation in somatic human tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 18018–18023. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Mariani, J.; Palejev, D.; Zhang, Y.; Haney, M.S.; Tomasini, L.; Ferrandino, A.F.; Rosenberg Belmaker, L.A.; Szekely, A.; Wilson, M.; et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012, 492, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Rehen, S.K.; McConnell, M.J.; Kaushal, D.; Kingsbury, M.A.; Yang, A.H.; Chun, J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. USA 2001, 98, 13361–13366. [Google Scholar] [CrossRef] [PubMed]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; de Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by l1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Kalousek, D.K.; Vekemans, M. Confined placental mosaicism. J. Med. Genet. 1996, 33, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kalousek, D.K.; Dill, F.J. Chromosomal mosaicism confined to the placenta in human conceptions. Science 1983, 221, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Z.; Chiu, R.W.K.; Sun, H.; Akolekar, R.; Chan, K.C.A.; Leung, T.Y.; Jiang, P.; Zheng, Y.W.L.; Lun, F.M.F.; Chan, L.Y.S.; et al. Noninvasive prenatal diagnosis of fetal trisomy 18 and trisomy 13 by maternal plasma DNA sequencing. PLOS ONE 2011, 6, e21791. [Google Scholar] [CrossRef] [PubMed]
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002, 3, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Tanaka, F.; Yamamoto, M.; Doyu, M.; Nagamatsu, M.; Riku, S.; Mitsuma, T.; Sobue, G. Somatic mosaicism of the expanded cag trinucleotide repeat in mrnas for the responsible gene of machado-joseph disease (MJD), dentatorubral-pallidoluysian atrophy (DRPLA), and spinal and bulbar muscular atrophy (SBMA). Neurochem. Res. 1998, 23, 25–32. [Google Scholar] [CrossRef] [PubMed]
- James, C.D.; Carlbom, E.; Nordenskjold, M.; Collins, V.P.; Cavenee, W.K. Mitotic recombination of chromosome 17 in astrocytomas. Proc. Natl. Acad. Sci. USA 1989, 86, 2858–2862. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, A.; Fryns, J.P.; Van den Berghe, H. On the variable effect of mosaic normal/balanced chromosomal rearrangements in man. J. Med. Genet. 1990, 27, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Kotzot, D.; Schmitt, S.; Bernasconi, F.; Robinson, W.P.; Lurie, I.W.; Ilyina, H.; Méhes, K.; Hamel, B.C.J.; Otten, B.J.; Hergersberg, M.; et al. Uniparental disomy 7 in silver-russell syndrome and primordial growth retardation. Hum. Mol. Genet. 1995, 4, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Santiago, B.; Malats, N.; Rothman, N.; Armengol, L.; Garcia-Closas, M.; Kogevinas, M.; Villa, O.; Hutchinson, A.; Earl, J.; Marenne, G.; et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010, 87, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Slatter, R.E.; Elliott, M.; Welham, K.; Carrera, M.; Schofield, P.N.; Barton, D.E.; Maher, E.R. Mosaic uniparental disomy in beckwith-wiedemann syndrome. J. Med. Genet. 1994, 31, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Zori, R.T.; Gray, B.A.; Bent-Williams, A.; Driscoll, D.J.; Williams, C.A.; Zackowski, J.L. Preaxial acrofacial dysostosis (nager syndrome) associated with an inherited and apparently balanced X;9 translocation: Prenatal and postnatal late replication studies. Am. J. Med. Genet. 1993, 46, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.; Cepko, C.L. Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science 1992, 255, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Pleasure, S.J.; Anderson, S.; Hevner, R.; Bagri, A.; Marin, O.; Lowenstein, D.H.; Rubenstein, J.L. Cell migration from the ganglionic eminences is required for the development of hippocampal gabaergic interneurons. Neuron 2000, 28, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Leibrock, J.; Bailey, K.; Barde, Y.-A. Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature 1990, 344, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R. Growth control of nerve cells by a protein factor and its antiserum: Discovery of this factor may provide new leads to understanding of some neurogenetic processes. Science 1964, 143, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Kurek, K.C.; Luks, V.L.; Ayturk, U.M.; Alomari, A.I.; Fishman, S.J.; Spencer, S.A.; Mulliken, J.B.; Bowen, M.E.; Yamamoto, G.L.; Kozakewich, H.P.; et al. Somatic mosaic activating mutations in PIK3CA cause cloves syndrome. Am. J. Hum. Genet. 2012, 90, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Campbell, P.J. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 2014, 30, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Crotwell, P.L.; Hoyme, H.E. Advances in whole-genome genetic testing: From chromosomes to microarrays. Curr. Probl. Pediatr. Adolesc. Health Care 2012, 42, 47–73. [Google Scholar] [CrossRef] [PubMed]
- Bushman, D.M.; Chun, J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin. Cell Dev. Biol. 2013, 24, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Notini, A.J.; Craig, J.M.; White, S.J. Copy number variation and mosaicism. Cytogenet. Genome Res. 2008, 123, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Human interphase chromosomes: A review of available molecular cytogenetic technologies. Mol. Cytogenet. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Imataka, G.; Arisaka, O. Chromosome analysis using spectral karyotyping (sky). Cell Biochem. Biophys. 2012, 62, 13–17. [Google Scholar] [PubMed]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Albertson, D.G. Array comparative genomic hybridization and its applications in cancer. Nat. Genet. 2005, 37, S11–S17. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Huang, J.; Greshock, J.; Watt, S.; Butler, A.; West, S.; Grigorova, M.; Jones, K.W.; Wei, W.; Stratton, M.R.; et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004, 14, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Leikauf, G.D.; Keith, G.; Rihn, B.H. Microarrays as cancer keys: An array of possibilities. J. Clin. Oncol. 2002, 20, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Baugher, J.D.; Baugher, B.D.; Shirley, M.D.; Pevsner, J. Sensitive and specific detection of mosaic chromosomal abnormalities using the parent-of-origin-based detection (POD) method. BMC Genom. 2013, 14, 367. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.K.; Chang, X.; Curtis, C.; Wang, K. Precise inference of copy number alterations in tumor samples from SNP arrays. Bioinformatics 2013, 29, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, Z.; Lezon-Geyda, K.; Sarkar, S.; Lannin, D.; Schulz, V.; Krop, I.; Winer, E.; Harris, L.; Tuck, D. GPHMM: An integrated hidden Markov model for identification of copy number alteration and loss of heterozygosity in complex tumor samples using whole genome SNP arrays. Nucleic Acids Res. 2011, 39, 4928–4941. [Google Scholar] [CrossRef]
- Liu, Z.; Li, A.; Schulz, V.; Chen, M.; Tuck, D. Mixhmm: Inferring copy number variation and allelic imbalance using SNP arrays and tumor samples mixed with stromal cells. PLOS ONE 2010, 5, e10909. [Google Scholar] [CrossRef] [PubMed]
- Rancoita, P.; Hutter, M.; Bertoni, F.; Kwee, I. An integrated bayesian analysis of LOH and copy number data. BMC Bioinform. 2010, 11, 321. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The sanger FASTQ file format for sequences with quality scores, and the solexa/illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Özen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. 2009, 106, 19096–19101. [Google Scholar] [CrossRef]
- Hodges, E.; Xuan, Z.; Balija, V.; Kramer, M.; Molla, M.N.; Smith, S.W.; Middle, C.M.; Rodesch, M.J.; Albert, T.J.; Hannon, G.J.; et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007, 39, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. Somaticsniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Ding, J.; Morin, R.; Crisan, A.; Ha, G.; Giuliany, R.; Bashashati, A.; Hirst, M.; Turashvili, G.; Oloumi, A.; et al. Jointsnvmix: A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012, 28, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [PubMed]
- Amarasinghe, K.C.; Li, J.; Halgamuge, S.K. Convex: Copy number variation estimation in exome sequencing data using HMM. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-freec: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. Lumpy: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gunel, M.; Zhao, H. Somatica: Identifying, characterizing and quantifying somatic copy number aberrations from cancer genome sequencing data. PLOS ONE 2013, 8, e78143. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wendl, M.C.; McMichael, J.F.; Raphael, B.J. Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 2014, 15, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; De, S. An assessment of computational methods for estimating purity and clonality using genomic data derived from heterogeneous tumor tissue samples. Brief. Bioinform. 2014. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Hosono, S.; Faruqi, A.F.; Dean, F.B.; Du, Y.; Sun, Z.; Wu, X.; Du, J.; Kingsmore, S.F.; Egholm, M.; Lasken, R.S. Unbiased whole-genome amplification directly from clinical samples. Genome Res. 2003, 13, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Delaney, A.D.; Farnoud, N.; Flibotte, S.; Griffith, M.; Li, H.I.; Qian, H.; Farinha, P.; Gascoyne, R.D.; Marra, M.A. Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res. 2008, 36, e80. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Kendall, J.; Rodgers, L.; Cox, H.; Riggs, M.; Stepansky, A.; Troge, J.; Ravi, K.; Esposito, D.; Lakshmi, B.; et al. Genome-wide copy number analysis of single cells. Nat. Protoc. 2012, 7, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Gundry, M.; Li, W.; Maqbool, S.B.; Vijg, J. Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res. 2012, 40, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, L.; Zhu, P.; Zhang, B.; Tao, Y.; Xu, X.; Li, F.; Wu, K.; Liang, J.; Shao, D.; et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012, 148, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hou, Y.; Yin, X.; Bao, L.; Tang, A.; Song, L.; Li, F.; Tsang, S.; Wu, K.; Wu, H.; et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 2012, 148, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.M.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacoke, M.; et al. Germline mutations of the pten gene in cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.; Strong, L.; Fraumeni, J.; Nelson, C.; Kim, D.; Kassel, J.; Gryka, M.; Bischoff, F.; Tainsky, M.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-TCF signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, M.; Konishi, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Yasuno, M.; Igari, T.; Koike, M.; Chiba, M.; Mori, T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1997, 17, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968, 218, 652–656. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. Cosmic: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Collins, F.S.; Barker, A.D. Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies. Sci. Am. 2007, 296, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Debeljak, M.; Freed, D.N.; Welch, J.A.; Haley, L.; Beierl, K.; Iglehart, B.S.; Pallavajjala, A.; Gocke, C.D.; Leffell, M.S.; Lin, M.-T.; et al. Haplotype counting by next-generation sequencing for ultrasensitive human DNA detection. J. Mol. Diagn. 2014, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 1954, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Szilard, L. On the nature of the aging process. Proc. Natl. Acad. Sci. USA 1959, 45, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Curtis, H.J. Biological mechanisms underlying the aging process. Science 1963, 141, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Goldsby, R.E.; Hays, L.E.; Chen, X.; Olmsted, E.A.; Slayton, W.B.; Spangrude, G.J.; Preston, B.D. High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading. Proc. Natl. Acad. Sci. USA 2002, 99, 15560–15565. [Google Scholar] [CrossRef]
- Goldsby, R.E.; Lawrence, N.A.; Hays, L.E.; Olmsted, E.A.; Chen, X.; Singh, M.; Preston, B.D. Defective DNA polymerase-[delta] proofreading causes cancer susceptibility in mice. Nat. Med. 2001, 7, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Albertson, T.M.; Ogawa, M.; Bugni, J.M.; Hays, L.E.; Chen, Y.; Wang, Y.; Treuting, P.M.; Heddle, J.A.; Goldsby, R.E.; Preston, B.D. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17101–17104. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Bielas, J.H.; Kujoth, G.C.; Ladiges, W.C.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007, 39, 540–543. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Hickson, I.D. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum. Mol. Genet. 2001, 10, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.; Kidd, A.; Mehes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in bub1b. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Date, H.; Onodera, O.; Tanaka, H.; Iwabuchi, K.; Uekawa, K.; Igarashi, S.; Koike, R.; Hiroi, T.; Yuasa, T.; Awaya, Y.; et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001, 29, 184–188. [Google Scholar] [CrossRef]
- Moreira, M.-C.; Barbot, C.; Tachi, N.; Kozuka, N.; Uchida, E.; Gibson, T.; Mendonca, P.; Costa, M.; Barros, J.; Yanagisawa, T.; et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001, 29, 189–193. [Google Scholar] [CrossRef]
- Niedernhofer, L.J. Tissue-specific accelerated aging in nucleotide excision repair deficiency. Mech. Ageing Dev. 2008, 129, 408–415. [Google Scholar] [CrossRef]
- Monnat, R.J., Jr. Human RECQ helicases: Roles in DNA metabolism, mutagenesis and cancer biology. Semin. Cancer Biol. 2010, 20, 329–339. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar]
- Goldfarb, L.G.; Brown, P.; McCombie, W.R.; Goldgaber, D.; Swergold, G.D.; Wills, P.R.; Cervenakova, L.; Baron, H.; Gibbs, C.J.; Gajdusek, D.C. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad. Sci. USA 1991, 88, 10926–10930. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Uphill, J.B.; Adamson, G.; Geddes, J.F.; Revesz, T.; Davis, M.B.; Wood, N.W.; Collinge, J.; et al. Somatic and germline mosaicism in sporadic early-onset alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Alzualde, A.; Moreno, F.; Martínez-Lage, P.; Ferrer, I.; Gorostidi, A.; Otaegui, D.; Blázquez, L.; Atares, B.; Cardoso, S.; Martínez de Pancorbo, M.; et al. Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178n mutation in the PRNP gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1283–1291. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Bate, C.; Van Gool, W.A.; Hoozemans, J.J.M.; Rozemuller, J.M.; Veerhuis, R.; Williams, A. Neuroinflammation in alzheimer’s disease and prion disease. Glia 2002, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of beta-amyloid by intracerebral infusion of alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of aβ prions from alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Breteler, M.M.B.; van Harskamp, F.; Claus, J.J.; van der Cammen, T.J.M.; Grobbee, D.E.; Hofman, A. Prevalence of alzheimer’s disease and vascular dementia: Association with education. The Rotterdam study. BMJ 1995, 310, 970. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Ben-Israel, J.; Goldhammer, Y.; Korczyn, A.D. The risk of developing creutzfeldt-jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology 1994, 44, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The AKT-mtor tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Mirzaa, G.; Conaway, R.; Graham, J.M., Jr.; Dobyns, W.B. PIK3CA-related segmental overgrowth. In GeneReviews; Pagon, R.A., Adam, M.P., Ardinger, H.H., et al., Eds.; University of Washington Seattle: Seattle, WA, USA, 2013. [Google Scholar]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Riviere, J.B.; Mirzaa, G.M.; O'Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mtor pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. RHEB is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell. Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Henske, E.P.; Wessner, L.L.; Golden, J.; Scheithauer, B.W.; Vortmeyer, A.O.; Zhuang, Z.; Klein-Szanto, A.J.; Kwiatkowski, D.J.; Yeung, R.S. Loss of tuberin in both subependymal giant cell astrocytomas and angiomyolipomas supports a two-hit model for the pathogenesis of tuberous sclerosis tumors. Am. J. Pathol. 1997, 151, 1639–1647. [Google Scholar] [PubMed]
- Tsang, E.; Birch, P.; Friedman, J.M. Valuing gene testing in children with possible neurofibromatosis 1. Clin. Genet. 2012, 82, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in ollier disease and maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Jamuar, S.S.; Lam, A.T.; Kircher, M.; D’Gama, A.M.; Wang, J.; Barry, B.J.; Zhang, X.; Hill, R.S.; Partlow, J.N.; Rozzo, A.; et al. Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 2014, 371, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Choate, K.A.; Lu, Y.; Zhou, J.; Choi, M.; Elias, P.M.; Farhi, A.; Nelson-Williams, C.; Crumrine, D.; Williams, M.L.; Nopper, A.J.; et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science 2010, 330, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Pasmooij, A.M.; Jonkman, M.F.; Uitto, J. Revertant mosaicism in heritable skin diseases: Mechanisms of natural gene therapy. Discov. Med. 2012, 14, 167–179. [Google Scholar] [PubMed]
- Pasmooij, A.M.; Pas, H.H.; Bolling, M.C.; Jonkman, M.F. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J. Clin. Invest. 2007, 117, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, R.; Yang, D.R.; Puck, J.M.; Huie, M.L.; Jiang, C.K.; Kurlandsky, L.E. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat. Genet. 1996, 13, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Leblanc, T.; Larghero, J.; Dastot, H.; Shimamura, A.; Guardiola, P.; Esperou, H.; Ferry, C.; Jubert, C.; Feugeas, J.-P.; et al. Detection of somatic mosaicism and classification of fanconi anemia patients by analysis of the FA/BRCA pathway. Blood 2005, 105, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- De, S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011, 27, 217–223. [Google Scholar] [PubMed]
- Insel, T.R. Brain somatic mutations: The dark matter of psychiatric genetics [quest]. Mol. Psychiatry 2014, 19, 156–158. [Google Scholar] [PubMed]
- Krumm, N.; O’Roak, B.J.; Shendure, J.; Eichler, E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014, 37, 95–105. [Google Scholar] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freed, D.; Stevens, E.L.; Pevsner, J. Somatic Mosaicism in the Human Genome. Genes 2014, 5, 1064-1094. https://doi.org/10.3390/genes5041064
Freed D, Stevens EL, Pevsner J. Somatic Mosaicism in the Human Genome. Genes. 2014; 5(4):1064-1094. https://doi.org/10.3390/genes5041064
Chicago/Turabian StyleFreed, Donald, Eric L. Stevens, and Jonathan Pevsner. 2014. "Somatic Mosaicism in the Human Genome" Genes 5, no. 4: 1064-1094. https://doi.org/10.3390/genes5041064
APA StyleFreed, D., Stevens, E. L., & Pevsner, J. (2014). Somatic Mosaicism in the Human Genome. Genes, 5(4), 1064-1094. https://doi.org/10.3390/genes5041064