
Toward Precision Medicine: Molecular Biomarkers of Response to Tofacitinib in Inflammatory Bowel Disease


Abstract
1. Introduction
1.1. From Traditional Immunosuppressive Therapies to More Targeted Treatment Approaches of UC
1.2. Treatment Protocols of UC Management
2. Tofacitinib
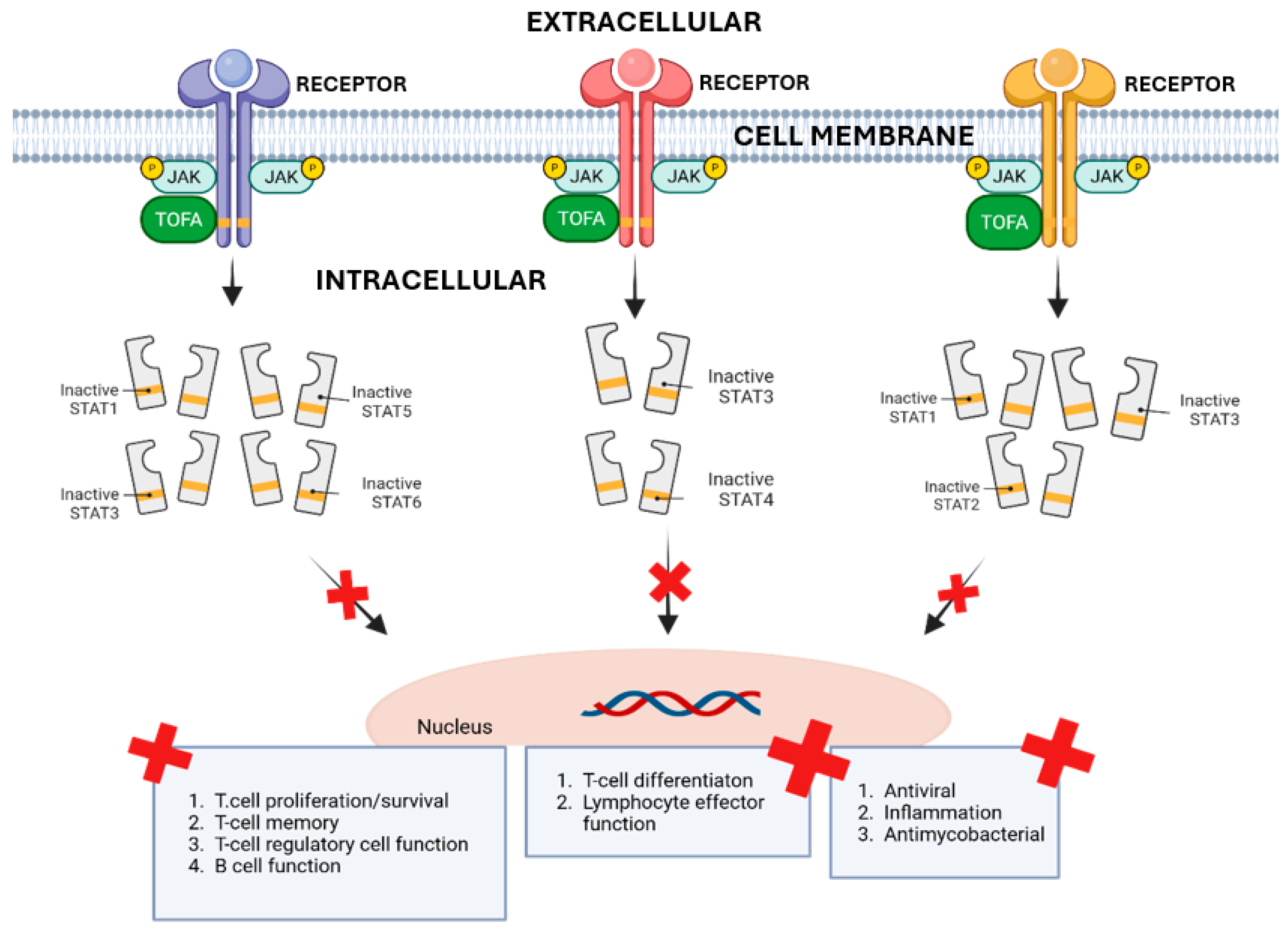
2.1. TOFA Mechanism and Development
2.2. TOFA Limitations
3. Epigenomics
Methylation Patterns in Response to TOFA Treatment
4. Transcriptomic
4.1. Modulation of Immune-Related Gene Expression by TOFA
4.2. Biomarkers of Tofacitinib Response: Responders vs. Non-Responders
4.3. Dual Role of TOFA at the Transcriptomic Layer
4.4. Potential Predictive Transcriptomic Biomarkers of Response to TOFA Treatment
4.5. The Role of miRNAs as Predictors of TOFA Treatment Response
5. Proteomics
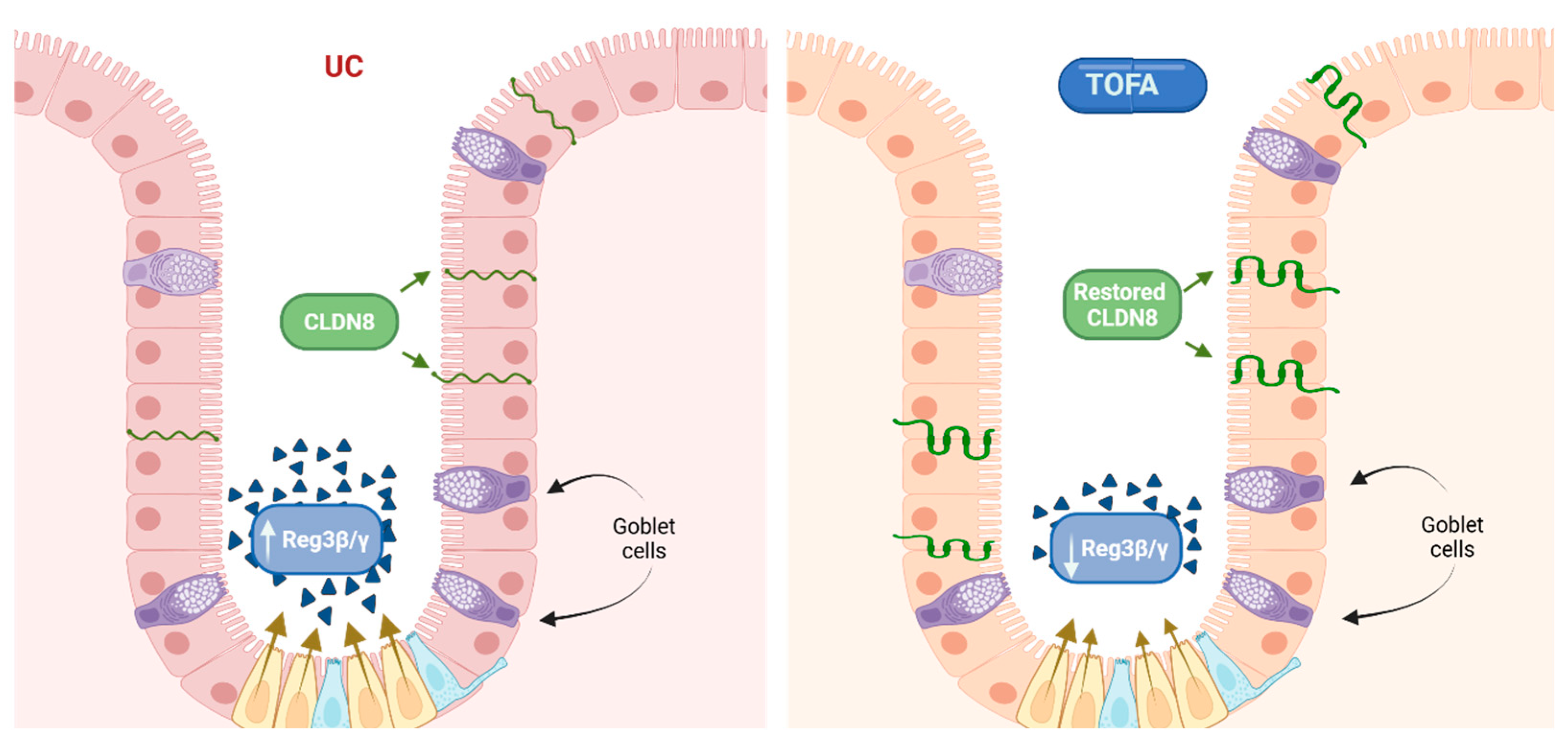
5.1. The Complexity of the TOFA Mechanism in Inflammation
5.2. Dual Role of TOFA at the Proteomic Layer
6. Cells as Biomarkers
6.1. Different Cell Biomarkers of Response to TOFA in Responders vs. Non-Responders
6.2. Cells as Predictors of Response to TOFA Treatment
7. Correlation of Epigenetically Altered Genes in UC with Biomarkers of TOFA Response
Evidence Supporting TOFA’s Dual Mechanism of Action
8. Future Challenges
8.1. Current Limitations in TOFA Treatment of UC
8.2. Unmet Needs and Challenges in Predictive Biomarkes to TOFA Treatment
8.3. Lack of miRNA Studies in Response to TOFA Treatment
8.4. TOFA Beyond Immunosuppression
9. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD | Crohn’s Disease |
| circRNA | Circular RNA |
| CpG | Cytosine-phosphate-Guanine site |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| GWASs | Genome-Wide Association Studies |
| IBD | Inflammatory Bowel Disease |
| lncRNA | Long Non-coding RNA |
| miRNA | MicroRNA |
| ncRNA | Non-coding RNA |
| NR | Non-Responder(s) |
| R | Responder(s) |
| RA | Rheumatoid Arthritis |
| scRNA-seq | Single-cell RNA sequencing |
| SNP | Single Nucleotide Polymorphism |
| Th1 | T-helper 1 cells |
| Th17 | T-helper 17 cells |
| TNF-α | Tumour Necrosis Factor Alpha |
| TOFA | Tofacitinib |
| UC | Ulcerative Colitis |
References
- Sinagra, E.; Utzeri, E.; Morreale, G.C.; Fabbri, C.; Pace, F.; Anderloni, A. Microbiota-gut-brain axis and its affect inflammatory bowel disease: Pathophysiological concepts and insights for clinicians. World J. Clin. Cases 2020, 8, 1013–1025. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- McDowell, C.; Farooq, U.; Haseeb, M. Inflammatory Bowel Disease. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2025. [Google Scholar]
- Abraham, B.P.; Ahmed, T.; Ali, T. Inflammatory Bowel Disease: Pathophysiology and Current Therapeutic Approaches. In Gastrointestinal Pharmacology; Greenwood-Van Meerveld, B., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 115–146. [Google Scholar]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Murch, S.H.; Lamkin, V.A.; Savage, M.O.; Walker-Smith, J.A.; MacDonald, T.T. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut 1991, 32, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Braegger, C.P.; Nicholls, S.; Murch, S.H.; MacDonald, T.T.; Stephens, S. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 1992, 339, 89–91. [Google Scholar] [CrossRef]
- Breese, E.J.; Michie, C.A.; Nicholls, S.W.; Murch, S.H.; Williams, C.B.; Domizio, P.; Walker-Smith, J.A.; Macdonald, T.T. Tumor necrosis factor α-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology 1994, 106, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Weber, C.R.; Schwarz, B.T.; Austin, J.R., 2nd; Raleigh, D.R.; Guan, Y.; Watson, A.J.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Ruder, B.; Atreya, R.; Becker, C. Tumour Necrosis Factor Alpha in Intestinal Homeostasis and Gut Related Diseases. Int. J. Mol. Sci. 2019, 20, 1887. [Google Scholar] [CrossRef] [PubMed]
- Derkx, B.; Taminiau, J.; Radema, S.; Stronkhorst, A.; Wortel, C.; Tytgat, G.; van Deventer, S. Tumour-necrosis-factor antibody treatment in Crohn’s disease. Lancet 1993, 342, 173–174. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Manrai, M.; Jha, A.A.; Dawra, S.; Pachisia, A.V. Biologics, Small Molecules and More in Inflammatory Bowel Disease: The Present and the Future. Future Pharmacol. 2024, 4, 279–316. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, Å.V. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease-Focusing on Intestinal Barrier Function. Cells 2019, 8, 193. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Feagan, B.G.; Panés, J.; Ferrante, M.; Kaser, A.; D’Haens, G.R.; Sandborn, W.J.; Louis, E.; Neurath, M.F.; Franchimont, D.; Dewit, O.; et al. Risankizumab in patients with moderate to severe Crohn’s disease: An open-label extension study. Lancet Gastroenterol. Hepatol. 2018, 3, 671–680. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Loftus, E.V., Jr.; Danese, S.; Colombel, J.F.; Törüner, M.; Jonaitis, L.; Abhyankar, B.; Chen, J.; Rogers, R.; et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Abreu, M.T.; D’Haens, G.; Colombel, J.F.; Vermeire, S.; Mitchev, K.; Jamoul, C.; Fedorak, R.N.; Spehlmann, M.E.; Wolf, D.C.; et al. Certolizumab Pegol in Patients with Moderate to Severe Crohn’s Disease and Secondary Failure to Infliximab. Clin. Gastroenterol. Hepatol. 2010, 8, 688–695.e2. [Google Scholar] [CrossRef]
- Bálint, A.; Farkas, K.; Palatka, K.; Lakner, L.; Miheller, P.; Rácz, I.; Hegede, G.; Vincze, Á.; Horváth, G.; Szabó, A.; et al. Efficacy and Safety of Adalimumab in Ulcerative Colitis Refractory to Conventional Therapy in Routine Clinical Practice. J. Crohn’s Colitis 2015, 10, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Marano, C.; Zhang, H.; Strauss, R.; Johanns, J.; Adedokun, O.J.; Guzzo, C.; Colombel, J.-F.; Reinisch, W.; et al. Subcutaneous Golimumab Induces Clinical Response and Remission in Patients with Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2014, 146, 85–95. [Google Scholar] [CrossRef]
- Greener, T.; Boland, K.; Steinhart, A.H.; Silverberg, M.S. The Unfinished Symphony: Golimumab Therapy for Anti-Tumour Necrosis Factor Refractory Crohn’s Disease. J. Crohn’s Colitis 2017, 12, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, K.; Gils, A.; Rutgeerts, P.; Levesque, B.G.; Vermeire, S.; Sandborn, W.J.; Vande Casteele, N. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: Evolution in the definition and management of primary nonresponse. Inflamm. Bowel Dis. 2015, 21, 182–197. [Google Scholar] [CrossRef]
- Schnitzler, F.; Fidder, H.; Ferrante, M.; Noman, M.; Arijs, I.; Van Assche, G.; Hoffman, I.; Van Steen, K.; Vermeire, S.; Rutgeerts, P. Long-term outcome of treatment with infliximab in 614 patients with Crohn’s disease: Results from a single-centre cohort. Gut 2009, 58, 492–500. [Google Scholar] [CrossRef]
- Long, D. Crohn’s Disease and Ulcerative Colitis: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2024, 12, 689. [Google Scholar] [CrossRef] [PubMed]
- Gorenjak, M.; Gole, B.; Goričan, L.; Jezernik, G.; Prosenc Zmrzljak, U.; Pernat, C.; Skok, P.; Potočnik, U. Single-Cell Transcriptomic and Targeted Genomic Profiling Adjusted for Inflammation and Therapy Bias Reveal CRTAM and PLCB1 as Novel Hub Genes for Anti-Tumor Necrosis Factor Alpha Therapy Response in Crohn’s Disease. Pharmaceutics 2024, 16, 835. [Google Scholar] [CrossRef]
- Roskos, L.K.; Ren, S.; Robbie, G. Application of Modeling and Simulation in the Development of Protein Drugs. In Clinical Trial Simulations: Applications and Trends; Kimko, H.H.C., Peck, C.C., Eds.; Springer: New York, NY, USA, 2011; pp. 361–396. [Google Scholar]
- Chanchlani, N.; Lin, S.; Bewshea, C.; Hamilton, B.; Thomas, A.; Smith, R.; Roberts, C.; Bishara, M.; Nice, R.; Lees, C.W.; et al. Mechanisms and management of loss of response to anti-TNF therapy for patients with Crohn’s disease: 3-year data from the prospective, multicentre PANTS cohort study. Lancet Gastroenterol. Hepatol. 2024, 9, 521–538. [Google Scholar] [CrossRef]
- Andersen, N.N.; Jess, T. Risk of infections associated with biological treatment in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 16014–16019. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Sharara, A.I.; Su, C.; Modesto, I.; Mundayat, R.; Gunay, L.M.; Salese, L.; Sands, B.E. Efficacy and Safety of Tofacitinib in Ulcerative Colitis Based on Prior Tumor Necrosis Factor Inhibitor Failure Status. Clin. Gastroenterol. Hepatol. 2022, 20, 591–601.e8. [Google Scholar] [CrossRef]
- Sands, B.E.; D’Haens, G.; Panaccione, R.; Regueiro, M.; Ghosh, S.; Hudesman, D.; Ahmad, H.A.; Mehra, D.; Wu, H.; Jain, A.; et al. Ozanimod in Patients with Moderate to Severe Ulcerative Colitis Naive to Advanced Therapies. Clin. Gastroenterol. Hepatol. 2024, 22, 2084–2095.e4. [Google Scholar] [CrossRef] [PubMed]
- Hvisdas, C.; Gleeson, P.K.; Apter, A.J. Cost considerations for clinicians prescribing biologic drugs: Who pays? J. Allergy Clin. Immunol. 2020, 146, 266–269. [Google Scholar] [CrossRef]
- Makurvet, F.D. Biologics vs. small molecules: Drug costs and patient access. Med. Drug Discov. 2021, 9, 100075. [Google Scholar] [CrossRef]
- Straatmijer, T.; Biemans, V.B.C.; Visschedijk, M.; Hoentjen, F.; de Vries, A.; van Bodegraven, A.A.; Bodelier, A.; de Boer, N.K.H.; Dijkstra, G.; Festen, N.; et al. Superior Effectiveness of Tofacitinib Compared to Vedolizumab in Anti-TNF-experienced Ulcerative Colitis Patients: A Nationwide Dutch Registry Study. Clin. Gastroenterol. Hepatol. 2023, 21, 182–191.e2. [Google Scholar] [CrossRef] [PubMed]
- Malakar, S.; Kothalkar, S.; Shamsul Hoda, U.; Ghoshal, U.C. Tofacitinib in Steroid-Refractory Acute Severe Ulcerative Colitis: A Retrospective Analysis. Cureus 2023, 15, e45416. [Google Scholar] [CrossRef]
- Taylor, P.C.; Choy, E.; Baraliakos, X.; Szekanecz, Z.; Xavier, R.M.; Isaacs, J.D.; Strengholt, S.; Parmentier, J.M.; Lippe, R.; Tanaka, Y. Differential properties of Janus kinase inhibitors in the treatment of immune-mediated inflammatory diseases. Rheumatology 2024, 63, 298–308. [Google Scholar] [CrossRef]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting Specificity in the Janus Kinases: The Structures of JAK-Specific Inhibitors Complexed to the JAK1 and JAK2 Protein Tyrosine Kinase Domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef]
- Müller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature 1993, 366, 129–135. [Google Scholar] [CrossRef]
- Lai, K.S.; Jin, Y.; Graham, D.K.; Witthuhn, B.A.; Ihle, J.N.; Liu, E.T. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J. Biol. Chem. 1995, 270, 25028–25036. [Google Scholar] [CrossRef]
- Wilks, A.F.; Harpur, A.G.; Kurban, R.R.; Ralph, S.J.; Zürcher, G.; Ziemiecki, A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol. Cell. Biol. 1991, 11, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Bonelli, M.; Gadina, M.; O’Shea, J.J. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat. Rev. Rheumatol. 2016, 12, 25–36. [Google Scholar] [CrossRef]
- Boland, B.S.; Sandborn, W.J.; Chang, J.T. Update on Janus Kinase Antagonists in Inflammatory Bowel Disease. Gastroenterol. Clin. N. Am. 2014, 43, 603–617. [Google Scholar] [CrossRef]
- Villarino, A.V.; Gadina, M.; O’Shea, J.J.; Kanno, Y. SnapShot: Jak-STAT Signaling II. Cell 2020, 181, 1696–1696.e1. [Google Scholar] [CrossRef]
- Flanagan, M.E.; Blumenkopf, T.A.; Brissette, W.H.; Brown, M.F.; Casavant, J.M.; Shang-Poa, C.; Doty, J.L.; Elliott, E.A.; Fisher, M.B.; Hines, M.; et al. Discovery of CP-690,550: A Potent and Selective Janus Kinase (JAK) Inhibitor for the Treatment of Autoimmune Diseases and Organ Transplant Rejection. J. Med. Chem. 2010, 53, 8468–8484. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J.; Blumenthal, D.E. Protein kinase small molecule inhibitors for rheumatoid arthritis: Medicinal chemistry/clinical perspectives. World J. Orthop. 2014, 5, 496–503. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Vande Casteele, N. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Inc. Pfizer Announces U.S. FDA Approves XELJANZ® (Tofacitinib) for the Treatment of Moderately to Severely Active Ulcerative Colitis. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_u_s_fda_approves_xeljanz_tofacitinib_for_the_treatment_of_moderately_to_severely_active_ulcerative_colitis-0?utm_source=chatgpt.com (accessed on 14 May 2025).
- Pfizer Inc. XELJANZ® (Tofacitinib Citrate) Receives Marketing Authorization in the European Union for Moderately to Severely Active Ulcerative Colitis. Available online: https://www.pfizer.com/news/press-release/press-release-detail/xeljanz_tofacitinib_citrate_receives_marketing_authorization_in_the_european_union_for_moderately_to_severely_active_ulcerative_colitis-0?utm_source=chatgpt.com (accessed on 14 May 2025).
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2025 update. Pharmacol. Res. 2025, 216, 107723. [Google Scholar] [CrossRef]
- Singh, A.; Goyal, M.K.; Midha, V.; Mahajan, R.; Kaur, K.; Gupta, Y.K.; Singh, D.; Bansal, N.; Kaur, R.; Kalra, S.; et al. Tofacitinib in Acute Severe Ulcerative Colitis (TACOS): A Randomized Controlled Trial. Am. J. Gastroenterol. 2024, 119, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Lawendy, N.; Danese, S.; Su, C.; Loftus, E.V., Jr.; Hart, A.; Dotan, I.; Damião, A.; Judd, D.T.; Guo, X.; et al. Safety and efficacy of tofacitinib for treatment of ulcerative colitis: Final analysis of OCTAVE Open, an open-label, long-term extension study with up to 7.0 years of treatment. Aliment. Pharmacol. Ther. 2022, 55, 464–478. [Google Scholar] [CrossRef]
- Khan, N.; Sundararajan, R.; Patel, M.; Trivedi, C.; Yang, Y.-X. Effectiveness of Tofacitinib in Patients With Ulcerative Colitis: A Nationwide Veterans Administration Cohort Study. Am. J. Gastroenterol. 2024, 119, 1632–1635. [Google Scholar] [CrossRef]
- Viola, A.; Li Voti, R.; Bivacqua, C.; De Francesco, C.; Muscianisi, M.; Costantino, G.; Fries, W. Mitigating the Risk of Tofacitinib-induced Adverse Events in the Elderly Population with Ulcerative Colitis. J. Crohn’s Colitis 2024, 18, 488–491. [Google Scholar] [CrossRef]
- Shimizu, H.; Aonuma, Y.; Hibiya, S.; Kawamoto, A.; Takenaka, K.; Fujii, T.; Saito, E.; Nagahori, M.; Ohtsuka, K.; Okamoto, R. Long-term efficacy and safety of tofacitinib in patients with ulcerative colitis: 3-year results from a real-world study. Intest. Res. 2024, 22, 369–377. [Google Scholar] [CrossRef]
- Shin, S.H.; Oh, K.; Hong, S.N.; Lee, J.; Oh, S.J.; Kim, E.S.; Na, S.Y.; Kang, S.B.; Koh, S.J.; Bang, K.B.; et al. Real-life effectiveness and safety of tofacitinib treatment in patients with ulcerative colitis: A KASID multicenter cohort study. Ther. Adv. Gastroenterol. 2023, 16, 17562848231154103. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Fujii, T.; Hibiya, S.; Motobayashi, M.; Suzuki, K.; Takenaka, K.; Saito, E.; Nagahori, M.; Ohtsuka, K.; Watanabe, M. Rapid prediction of 1-year efficacy of tofacitinib for treating refractory ulcerative colitis. Intest. Res. 2021, 19, 115–118. [Google Scholar] [CrossRef]
- Melón-Ardanaz, E.; Veny, M.; Corraliza, A.M.; Gudiño, V.; Garrido-Trigo, A.; Sanzo-Machuca, Á.; Buendia, M.; Esteller, M.; Robbins-Moreno, L.; Rodrigo, M.; et al. Differential effects of tofacitinib on macrophage activation contribute to lack of response in ulcerative colitis patients. J. Crohn’s Colitis 2025, 19, jjaf076. [Google Scholar] [CrossRef]
- Panés, J.; D’Haens, G.R.; Sands, B.E.; Ng, S.C.; Lawendy, N.; Kulisek, N.; Guo, X.; Wu, J.; Vranic, I.; Panaccione, R.; et al. Analysis of tofacitinib safety in ulcerative colitis from the completed global clinical developmental program up to 9.2 years of drug exposure. United Eur. Gastroenterol. J. 2024, 12, 793–801. [Google Scholar] [CrossRef]
- Mahadevan, U.; Baumgart, D.C.; Dubinsky, M.C.; Yamamoto-Furusho, J.K.; Lawendy, N.; Konijeti, G.G.; Gröchenig, H.P.; Jones, T.V.; Kulisek, N.; Kwok, K.; et al. S0847 Pregnancy Outcomes in the Tofacitinib Ulcerative Colitis OCTAVE Studies: An Update as of February 2020. Am. J. Gastroenterol. 2020, 115, S437–S438. [Google Scholar] [CrossRef]
- Panés, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; Haens, G.; Panaccione, R.; Higgins, P.D.R.; Colombel, J.-F.; Feagan, B.G.; et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: Results of two phase IIb randomised placebo-controlled trials. Gut 2017, 66, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Gordon, H.; Trier Moller, F.; Andersen, V.; Harbord, M. Heritability in inflammatory bowel disease: From the first twin study to genome-wide association studies. Inflamm. Bowel Dis. 2015, 21, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Loddo, I.; Romano, C. Inflammatory Bowel Disease: Genetics, Epigenetics, and Pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef]
- Banerjee, R.; Pal, P.; Prakash, N.; Mudigonda, S.; Joseph, S.; Patel, R.; Khalil, M.; Komawar, A.; Korikana, S.; Mekala, D.; et al. DOP60 Environmental risk factors for Inflammatory bowel disease: A large prospective case-control study in India. J. Crohn’s Colitis 2023, 17, i132–i135. [Google Scholar] [CrossRef]
- Sanmarco, L.M.; Chao, C.-C.; Wang, Y.-C.; Kenison, J.E.; Li, Z.; Rone, J.M.; Rejano-Gordillo, C.M.; Polonio, C.M.; Gutierrez-Vazquez, C.; Piester, G.; et al. Identification of environmental factors that promote intestinal inflammation. Nature 2022, 611, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Taman, H.; Fenton, C.G.; Hensel, I.V.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. Genome-Wide DNA Methylation in Treatment-Naïve Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Taman, H.; Fenton, C.G.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. DNA hypo-methylation facilitates anti-inflammatory responses in severe ulcerative colitis. PLoS ONE 2021, 16, e0248905. [Google Scholar] [CrossRef]
- Joustra, V.; Li Yim, A.Y.F.; van Gennep, S.; Hageman, I.; de Waard, T.; Levin, E.; Lauffer, P.; de Jonge, W.; Henneman, P.; Löwenberg, M.; et al. Peripheral Blood DNA Methylation Signatures and Response to Tofacitinib in Moderate-to-severe Ulcerative Colitis. J. Crohn’s Colitis 2024, 18, 1179–1189. [Google Scholar] [CrossRef]
- Nair, J.S.; DaFonseca, C.J.; Tjernberg, A.; Sun, W.; Darnell, J.E., Jr.; Chait, B.T.; Zhang, J.J. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-gamma. Proc. Natl. Acad. Sci. USA 2002, 99, 5971–5976. [Google Scholar] [CrossRef]
- Citores, L.; Bai, L.; Sørensen, V.; Olsnes, S. Fibroblast growth factor receptor-induced phosphorylation of STAT1 at the Golgi apparatus without translocation to the nucleus. J. Cell. Physiol. 2007, 212, 148–156. [Google Scholar] [CrossRef]
- Dezelak, M.; Repnik, K.; Koder, S.; Ferkolj, I.; Potocnik, U. A Prospective Pharmacogenomic Study of Crohn’s Disease Patients during Routine Therapy with Anti-TNF-alpha Drug Adalimumab: Contribution of ATG5, NFKB1, and CRP Genes to Pharmacodynamic Variability. OMICS 2016, 20, 296–309. [Google Scholar] [CrossRef]
- Gorenjak, M.; Repnik, K.; Jezernik, G.; Jurgec, S.; Skok, P.; Potocnik, U. Genetic prediction profile for adalimumab response in Slovenian Crohn’s disease patients. Z. Gastroenterol. 2019, 57, 1218–1225. [Google Scholar] [CrossRef]
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.K.; Potocnik, U. Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Repnik, K.; Koder, S.; Skok, P.; Ferkolj, I.; Potocnik, U. Transferrin Level Before Treatment and Genetic Polymorphism in HFE Gene as Predictive Markers for Response to Adalimumab in Crohn’s Disease Patients. Biochem. Genet. 2016, 54, 476–486. [Google Scholar] [CrossRef]
- Song, E.M.; Saqib, J.; Joo, Y.H.; Ramsha, Z.; Moon, C.M.; Jung, S.A.; Kim, J. Inflammatory transcriptomic signatures and cell type compositions in inflamed and non-inflamed colonic mucosa of ulcerative colitis. Genes Dis. 2025, 12, 101447. [Google Scholar] [CrossRef]
- Yu, K.; Peng, H.; Zhang, Z.; Ye, L.; Zhan, K.; Li, C.; Gan, L.; Lin, Y.; Wang, Y.; Song, Y.; et al. Long non-coding RNA ANRIL/p65 negative feedback loop protects intestinal barrier function in inflammatory bowel disease. Noncoding RNA Res. 2025, 12, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Taman, H.; Fenton, C.G.; Hensel, I.V.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. Transcriptomic Landscape of Treatment-Naïve Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 327–336. [Google Scholar] [CrossRef]
- Flood, P.; Fanning, A.; Woznicki, J.A.; Crowley, T.; Christopher, A.; Vaccaro, A.; Houston, A.; McSweeney, S.; Ross, S.; Hogan, A.; et al. DNA sensor-associated type I interferon signaling is increased in ulcerative colitis and induces JAK-dependent inflammatory cell death in colonic organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G439–G460. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef]
- Ito, Y.; Watanabe, D.; Okamoto, N.; Miyazaki, H.; Tokunaga, E.; Ku, Y.; Ooi, M.; Hoshi, N.; Kohashi, M.; Kanzawa, M.; et al. Activated type 17 helper T cells affect tofacitinib treatment outcomes. Sci. Rep. 2025, 15, 6112. [Google Scholar] [CrossRef]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S. Macrophage-Derived IGF-1 Is a Potent Coordinator of Myogenesis and Inflammation in Regenerating Muscle. Mol. Ther. 2015, 23, 1134–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, Z.M. Multiple functions of Maf in the regulation of cellular development and differentiation. Diabetes Metab. Res. Rev. 2015, 31, 773–778. [Google Scholar] [CrossRef]
- Shah, K.; Maradana, M.R.; Joaquina Delàs, M.; Metidji, A.; Graelmann, F.; Llorian, M.; Chakravarty, P.; Li, Y.; Tolaini, M.; Shapiro, M.; et al. Cell-intrinsic Aryl Hydrocarbon Receptor signalling is required for the resolution of injury-induced colonic stem cells. Nat. Commun. 2022, 13, 1827. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Balaji, S.; Le, L.D.; Crombleholme, T.M.; Keswani, S.G. Regenerative Wound Healing: The Role of Interleukin-10. Adv. Wound Care 2014, 3, 315–323. [Google Scholar] [CrossRef]
- Gonneaud, A.; Turgeon, N.; Boisvert, F.-M.; Boudreau, F.; Asselin, C. JAK-STAT Pathway Inhibition Partially Restores Intestinal Homeostasis in Hdac1- and Hdac2-Intestinal Epithelial Cell-Deficient Mice. Cells 2021, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.K.; Ercelen, D.; Cen Feng, J.Y.C.; Gurunathan, S.; Zhou, C.; Korman, A.; Newell, L.; Hudesman, D.; Jones, D.R.; Loke, P.; et al. Tofacitinib uptake by patient-derived intestinal organoids predicts individual clinical responsiveness. bioRxiv 2024, 167, 1453–1456.e5. [Google Scholar] [CrossRef] [PubMed]
- Parkes, G.; Ungaro, R.C.; Danese, S.; Abreu, M.T.; Arenson, E.; Zhou, W.; Ilo, D.; Laroux, F.S.; Deng, H.; Sanchez Gonzalez, Y.; et al. Correlation of mucosal healing endpoints with long-term clinical and patient-reported outcomes in ulcerative colitis. J. Gastroenterol. 2023, 58, 990–1002. [Google Scholar] [CrossRef]
- Sommer, K.; Wiendl, M.; Müller, T.M.; Heidbreder, K.; Voskens, C.; Neurath, M.F.; Zundler, S. Intestinal Mucosal Wound Healing and Barrier Integrity in IBD-Crosstalk and Trafficking of Cellular Players. Front. Med. 2021, 8, 643973. [Google Scholar] [CrossRef]
- Parlato, M.; Charbit-Henrion, F.; Pan, J.; Romano, C.; Duclaux-Loras, R.; Le Du, M.H.; Warner, N.; Francalanci, P.; Bruneau, J.; Bras, M.; et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol. Med. 2018, 10, e8483. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef]
- Komeda, Y.; Sakurai, T.; Sakai, K.; Morita, Y.; Hashimoto, A.; Nagai, T.; Hagiwara, S.; Matsumura, I.; Nishio, K.; Kudo, M. Refractory case of ulcerative colitis with idiopathic thrombocytopenic purpura successfully treated by Janus kinase inhibitor tofacitinib: A case report. World J. Clin. Cases 2020, 8, 6389–6395. [Google Scholar] [CrossRef]
- van Beelen Granlund, A.; Østvik, A.E.; Brenna, Ø.; Torp, S.H.; Gustafsson, B.I.; Sandvik, A.K. REG gene expression in inflamed and healthy colon mucosa explored by in situ hybridisation. Cell Tissue Res. 2013, 352, 639–646. [Google Scholar] [CrossRef]
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. N Y Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Tchoupa, A.K.; Schuhmacher, T.; Hauck, C.R. Signaling by epithelial members of the CEACAM family—Mucosal docking sites for pathogenic bacteria. Cell Commun. Signal. 2014, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Artis, D.; Becker, C. The intestinal barrier: A pivotal role in health, inflammation, and cancer. Lancet Gastroenterol. Hepatol. 2025, 10, 573–592. [Google Scholar] [CrossRef]
- Smithson, J.E.; Warren, B.F.; Young, S.; Pigott, R.; Jewell, D.P. Heterogeneous expression of carcinoembryonic antigen in the normal colon and upregulation in active ulcerative colitis. J. Pathol. 1996, 180, 146–151. [Google Scholar] [CrossRef]
- Barnich, N.; Carvalho, F.A.; Glasser, A.L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Gonzalo, G.; Hanrahan, N.; Rossini, V.; Singh, R.; Ahern, M.; Kelleher, M.; Hill, S.; O’Sullivan, R.; Fanning, A.; Walsh, P.T.; et al. Regulation of CEACAM Family Members by IBD-Associated Triggers in Intestinal Epithelial Cells, Their Correlation to Inflammation and Relevance to IBD Pathogenesis. Front. Immunol. 2021, 12, 655960. [Google Scholar] [CrossRef]
- Motohashi, H.; Inui, K.-I. Multidrug and toxin extrusion family SLC47: Physiological, pharmacokinetic and toxicokinetic importance of MATE1 and MATE2-K. Mol. Asp. Med. 2013, 34, 661–668. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, H.; Day, C.S.; Bierbach, U. Platinum–Acridine Agents with High Activity in Cancers Expressing the Solute Carrier MATE1 (SLC47A1). ACS Med. Chem. Lett. 2023, 14, 1122–1128. [Google Scholar] [CrossRef]
- Batara, D.C.; Park, S.W.; Kim, H.J.; Choi, S.Y.; Ohn, T.; Choi, M.C.; Park, S.I.; Kim, S.H. Targeting the multidrug and toxin extrusion 1 gene (SLC47A1) sensitizes glioma stem cells to temozolomide. Am. J. Cancer Res. 2023, 13, 4021–4038. [Google Scholar] [PubMed]
- Li, P.; Huang, T.; Zou, Q.; Liu, D.; Wang, Y.; Tan, X.; Wei, Y.; Qiu, H. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 2019, 202, 3065–3075. [Google Scholar] [CrossRef]
- Reid, K.M.; Brown, G.C. LRPAP1 is released from activated microglia and inhibits microglial phagocytosis and amyloid beta aggregation. Front. Immunol. 2023, 14, 1286474. [Google Scholar] [CrossRef] [PubMed]
- Maßberg, D.; Hatt, H. Human Olfactory Receptors: Novel Cellular Functions Outside of the Nose. Physiol. Rev. 2018, 98, 1739–1763. [Google Scholar] [CrossRef]
- Flegel, C.; Manteniotis, S.; Osthold, S.; Hatt, H.; Gisselmann, G. Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS ONE 2013, 8, e55368. [Google Scholar] [CrossRef]
- Qiu, C.; Chen, Y.; Xia, H.; Duan, J.; Zhang, L.; Zhang, Y.; Chen, Z.; Zhang, L. Hsa_circ_0004662 Accelerates the Progression of Ulcerative Colitis via the microRNA-532/HMGB3 Signalling Axis. J. Cell. Mol. Med. 2025, 29, e70430. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, Y.; Li, F.; Wang, Y.; Yang, J.; Gong, H.; Wan, X.; Ouyang, M. Circular RNA HECTD1 Mitigates Ulcerative Colitis by Promoting Enterocyte Autophagy Via miR-182-5p/HuR Axis. Inflamm. Bowel Dis. 2021, 28, 273–288. [Google Scholar] [CrossRef]
- Fasseu, M.; Tréton, X.; Guichard, C.; Pedruzzi, E.; Cazals-Hatem, D.; Richard, C.; Aparicio, T.; Daniel, F.; Soulé, J.C.; Moreau, R.; et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS ONE 2010, 5, e13160. [Google Scholar] [CrossRef]
- Pathak, S.; Grillo, A.R.; Scarpa, M.; Brun, P.; D’Incà, R.; Nai, L.; Banerjee, A.; Cavallo, D.; Barzon, L.; Palù, G.; et al. MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp. Mol. Med. 2015, 47, e164. [Google Scholar] [CrossRef]
- Viennois, E.; Zhao, Y.; Han, M.K.; Xiao, B.; Zhang, M.; Prasad, M.; Wang, L.; Merlin, D. Serum miRNA signature diagnoses and discriminates murine colitis subtypes and predicts ulcerative colitis in humans. Sci. Rep. 2017, 7, 2520. [Google Scholar] [CrossRef] [PubMed]
- Brusnic, O.; Adrian, B.; Sorin-Radu, F.; Grama, B.; Sofonea, F.; Roman-Filip, C.; Roman-Filip, I.; Solomon, A.; Bîrsan, S.; Dura, H.; et al. Importance of Fecal Microbiota Transplantation and Molecular Regulation as Therapeutic Strategies in Inflammatory Bowel Diseases. Nutrients 2024, 16, 4411. [Google Scholar] [CrossRef] [PubMed]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.; Kiss, A.; Arató, A.; Sziksz, E.; et al. Role of Altered Expression of miR-146a, miR-155, and miR-122 in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef]
- Schniers, A.; Goll, R.; Pasing, Y.; Sørbye, S.W.; Florholmen, J.; Hansen, T. Ulcerative colitis: Functional analysis of the in-depth proteome. Clin. Proteom. 2019, 16, 4. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 Is the Key Effector Th2 Cytokine in Ulcerative Colitis That Affects Epithelial Tight Junctions, Apoptosis, and Cell Restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H.; Sato, N.; Mizuno, N.; Ikawa, Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun. Rev. 2022, 21, 103017. [Google Scholar] [CrossRef]
- Bogaert, S.; Laukens, D.; Peeters, H.; Melis, L.; Olievier, K.; Boon, N.; Verbruggen, G.; Vandesompele, J.; Elewaut, D.; De Vos, M. Differential mucosal expression of Th17-related genes between the inflamed colon and ileum of patients with inflammatory bowel disease. BMC Immunol. 2010, 11, 61. [Google Scholar] [CrossRef]
- Lethen, I.; Lechner-Grimm, K.; Gabel, M.; Knauss, A.; Atreya, R.; Neurath, M.F.; Weigmann, B. Tofacitinib Affects M1-like and M2-like Polarization and Tissue Factor Expression in Macrophages of Healthy Donors and IBD Patients. Inflamm. Bowel Dis. 2024, 30, 1151–1163. [Google Scholar] [CrossRef]
- Cordes, F.; Lenker, E.; Spille, L.J.; Weinhage, T.; Bettenworth, D.; Kessel, C.; Schmidt, H.H.; Foell, D.; Varga, G. Tofacitinib Reprograms Human Monocytes of IBD Patients and Healthy Controls Toward a More Regulatory Phenotype. Inflamm. Bowel Dis. 2020, 26, 391–406. [Google Scholar] [CrossRef]
- Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Annunziato, F. Th17 plasticity: Pathophysiology and treatment of chronic inflammatory disorders. Curr. Opin. Pharmacol. 2014, 17, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Salem, Y.; Yacov, N.; Kafri, P.; Propheta-Meiran, O.; Karni, A.; Maharshak, N.; Furer, V.; Elkayam, O.; Mendel, I. MOSPD2 regulates the activation state of αLβ2 integrin to control monocyte migration: Applicability for treatment of chronic inflammatory diseases. Immunol. Res. 2025, 73, 78. [Google Scholar] [CrossRef]
- Chapuy, L.; Bsat, M.; Rubio, M.; Sarkizova, S.; Therrien, A.; Bouin, M.; Orlicka, K.; Weber, A.; Soucy, G.; Villani, A.-C.; et al. IL-12 and Mucosal CD14+ Monocyte-Like Cells Induce IL-8 in Colonic Memory CD4+ T Cells of Patients with Ulcerative Colitis but not Crohn’s Disease. J. Crohn’s Colitis 2019, 14, 79–95. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Sridhar, A.; Bakke, I.; Gopalakrishnan, S.; Osoble, N.M.M.; Hammarqvist, E.P.; Pettersen, H.P.S.; Sandvik, A.K.; Østvik, A.E.; Hansen, M.D.; Bruland, T. Tofacitinib and budesonide treatment affect stemness and chemokine release in IBD patient-derived colonoids. Sci. Rep. 2025, 15, 3753. [Google Scholar] [CrossRef]
- Bao, W.; Lyu, J.; Feng, G.; Guo, L.; Zhao, D.; You, K.; Liu, Y.; Li, H.; Du, P.; Chen, D.; et al. Aloe emodin promotes mucosal healing by modifying the differentiation fate of enteroendocrine cells via regulating cellular free fatty acid sensitivity. Acta Pharm. Sin. B 2024, 14, 3964–3982. [Google Scholar] [CrossRef]
- Zheng, L.; Duan, S.L. Molecular regulation mechanism of intestinal stem cells in mucosal injury and repair in ulcerative colitis. World J. Gastroenterol. 2023, 29, 2380–2396. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, Z.; Pan, T.; Hu, H.; He, R.; Yi, M.; Sun, W.; Gao, R.; Wang, H.; Ma, X.; et al. RNF186/EPHB2 Axis Is Essential in Regulating TNF Signaling for Colorectal Tumorigenesis in Colorectal Epithelial Cells. J. Immunol. 2022, 209, 1796–1805. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chu, S.; Liu, X.; Li, J.; Chen, Q.; Xu, M.; Wu, H.; Li, M.; Dong, Y.; Zhu, F.; et al. Extracellular vesicles derived from EphB2-overexpressing bone marrow mesenchymal stem cells ameliorate DSS-induced colitis by modulating immune balance. Stem Cell Res. Ther. 2021, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.M.; Moore, B.A.; Zyhowski, A.; Siegel, A.; Uttam, S.; Metter, E.J.; Engstrom, J.; Brand, R.E.; Biswas, N.; Whitcomb, D.C.; et al. Tofacitinib inhibits inflammatory cytokines from ulcerative colitis and healthy mucosal explants and is associated with pSTAT1/3 reduction in T-cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G396–G410. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tian, H.; Jiang, H.J.; Han, B. Interleukin-17 SNPs and serum levels increase ulcerative colitis risk: A meta-analysis. World J. Gastroenterol. 2014, 20, 15899–15909. [Google Scholar] [CrossRef]
- Krawiec, P.; Pac-Kożuchowska, E. Serum interleukin 17A and interleukin 17F in children with inflammatory bowel disease. Sci. Rep. 2020, 10, 12617. [Google Scholar] [CrossRef]
- Ito, R.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Kita, M.; Ueda, Y.; Iwakura, Y.; et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin. Exp. Immunol. 2006, 146, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Alhendi, A.; Naser, S.A. The dual role of interleukin-6 in Crohn’s disease pathophysiology. Front. Immunol. 2023, 14, 1295230. [Google Scholar] [CrossRef]
- Melón-Ardanaz, E.; Veny, M.; Corraliza, A.M.; Garrido-Trigo, A.; Esteller, M.; Rodrigo, M.; Verstockt, B.; Vermeire, S.; Masamunt, M.C.; Giner, Á.; et al. DOP05 Single-cell RNAseq temporal analysis of ulcerative colitis patients undergoing tofacitinib treatment reveals a shift in myeloid cells towards pro-inflammatory phenotypes in refractory patients. J. Crohn’s Colitis 2023, 17, i62–i64. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef]
- Sun, Z.; Ye, J.; Sun, W.; Jiang, L.; Shan, B.; Zhang, M.; Xu, J.; Li, W.; Liu, J.; Jing, H.; et al. Cooperation of TRADD- and RIPK1-dependent cell death pathways in maintaining intestinal homeostasis. Nat. Commun. 2025, 16, 1890. [Google Scholar] [CrossRef] [PubMed]
- Zan, G.X.; Qu, H.Z.; Li, X.Y.; Peng, Q.L.; Wang, X.F.; Li, R.S.; Zhao, Y.G.; Yan, H.C.; Zhou, J.Y.; Wang, X.Q. Iturin A Potentiates Differentiation of Intestinal Epithelial Defense Cells by Modulating Keap1/Nrf2 Signaling to Mitigate Oxidative Damage Induced by Heat-Stable Enterotoxin B. Antioxidants 2025, 14, 478. [Google Scholar] [CrossRef]
- Martín-Adrados, B.; Wculek, S.K.; Fernández-Bravo, S.; Torres-Ruiz, R.; Valle-Noguera, A.; Gomez-Sánchez, M.J.; Hernández-Walias, J.C.; Ferreira, F.M.; Corraliza, A.M.; Sancho, D.; et al. Expression of HMGCS2 in intestinal epithelial cells is downregulated in inflammatory bowel disease associated with endoplasmic reticulum stress. Front. Immunol. 2023, 14, 1185517. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta (BBA)–Mol. Cell Res. 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- Tamulytė, R.; Jankaitytė, E.; Toleikis, Z.; Smirnovas, V.; Jankunec, M. Pro-inflammatory protein S100A9 alters membrane organization by dispersing ordered domains. Biochim. Biophys. Acta (BBA)–Biomembr. 2023, 1865, 184113. [Google Scholar] [CrossRef]
- Aggeletopoulou, I.; Kalafateli, M.; Tsounis, E.P.; Triantos, C. Exploring the role of IL-1β in inflammatory bowel disease pathogenesis. Front. Med. 2024, 11, 1307394. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Sadanaga, T.; Nanni, L.; Wang, S.; Mizoguchi, A. Recently Updated Role of Chitinase 3-like 1 on Various Cell Types as a Major Influencer of Chronic Inflammation. Cells 2024, 13, 678. [Google Scholar] [CrossRef]
- Wang, N.; Liu, C.; Wang, X.; He, T.; Li, L.; Liang, X.; Wang, L.; Song, L.; Wei, Y.; Wu, Q.; et al. Hyaluronic Acid Oligosaccharides Improve Myocardial Function Reconstruction and Angiogenesis against Myocardial Infarction by Regulation of Macrophages. Theranostics 2019, 9, 1980–1992. [Google Scholar] [CrossRef]
- Tran, C.T.; Garcia, M.; Garnier, M.; Burucoa, C.; Bodet, C. Inflammatory signaling pathways induced by Helicobacter pylori in primary human gastric epithelial cells. Innate Immun. 2017, 23, 165–174. [Google Scholar] [CrossRef]
- Lee, H.-L.; Tsai, Y.-C.; Pikatan, N.W.; Yeh, C.-T.; Yadav, V.K.; Chen, M.-Y.; Tsai, J.-T. Tumor-Associated Macrophages Affect the Tumor Microenvironment and Radioresistance via the Upregulation of CXCL6/CXCR2 in Hepatocellular Carcinoma. Biomedicines 2023, 11, 2081. [Google Scholar] [CrossRef]
- Wang, X.; Dai, Y.; Zhang, X.; Pan, K.; Deng, Y.; Wang, J.; Xu, T. CXCL6 regulates cell permeability, proliferation, and apoptosis after ischemia-reperfusion injury by modulating Sirt3 expression via AKT/FOXO3a activation. Cancer Biol. Ther. 2021, 22, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Koga, H.; Tsuji, K.; Miyatake, K.; Nakagawa, Y.; Yokota, T.; Sekiya, I.; Katagiri, H. Extracellular vesicles derived from mesenchymal stromal cells mediate endogenous cell growth and migration via the CXCL5 and CXCL6/CXCR2 axes and repair menisci. Stem Cell Res. Ther. 2021, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, K.; Mosli, M.H.; Parker, C.E.; MacDonald, J.K. The impact of biological interventions for ulcerative colitis on health-related quality of life. Cochrane Database Syst. Rev. 2015, 2015, CD008655. [Google Scholar] [CrossRef] [PubMed]
- Paschos, P.; Katsoula, A.; Salanti, G.; Giouleme, O.; Athanasiadou, E.; Tsapas, A. Systematic review with network meta-analysis: The impact of medical interventions for moderate-to-severe ulcerative colitis on health-related quality of life. Aliment. Pharmacol. Ther. 2018, 48, 1174–1185. [Google Scholar] [CrossRef]
- Fine, S.; Papamichael, K.; Cheifetz, A.S. Etiology and Management of Lack or Loss of Response to Anti-Tumor Necrosis Factor Therapy in Patients with Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2019, 15, 656–665. [Google Scholar]
- Gisbert, J.P.; Panés, J. Loss of Response and Requirement of Infliximab Dose Intensification in Crohn’s Disease: A Review. Am. J. Gastroenterol. 2009, 104, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, A.; Nakashima, A.; Miyazaki, M.; Fujiki, M.; Kakimoto, H.; Hisabe, T.; Imakyure, O. Factors influencing the discontinuation of biologic therapies in patients with ulcerative colitis. J. Pharm. Health Care Sci. 2024, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, I.; Shahid, M.; Natesan, S.; Faruk, A.; Sood, A.K.; Khan, T. Hansen Solubility Parameters, Computational, and Thermodynamic Models for Tofacitinib Citrate Solubility in Neat Mono Solvents, and GastroPlus Based Predicted In Vivo Performance of Subcutaneous Solution in Humans. AAPS PharmSciTech 2025, 26, 64. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Panés, J.; Sands, B.E.; Reinisch, W.; Su, C.; Lawendy, N.; Koram, N.; Fan, H.; Jones, T.V.; Modesto, I.; et al. Venous thromboembolic events in the tofacitinib ulcerative colitis clinical development programme. Aliment. Pharmacol. Ther. 2019, 50, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Li Wai Suen, C.F.D.; Seah, D.; Choy, M.C.; De Cruz, P. Factors Associated with Response to Rescue Therapy in Acute Severe Ulcerative Colitis. Inflamm. Bowel Dis. 2024, 30, 1389–1405. [Google Scholar] [CrossRef]
- Effinger, A.; O’Driscoll, C.M.; McAllister, M.; Fotaki, N. Impact of gastrointestinal disease states on oral drug absorption—Implications for formulation design—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 674–698. [Google Scholar] [CrossRef]
- Lamba, V.; Panetta, J.C.; Strom, S.; Schuetz, E.G. Genetic predictors of interindividual variability in hepatic CYP3A4 expression. J. Pharmacol. Exp. Ther. 2010, 332, 1088–1099. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Gouju, J.; Legeay, S. Pharmacokinetics of obese adults: Not only an increase in weight. Biomed. Pharmacother. 2023, 166, 115281. [Google Scholar] [CrossRef] [PubMed]
- Sagawa, K.; Purohit, V.; Le, V.; Hsu, H.-J.; Dowty, M.E.; Tse, S.; Chang, C. Virtual Bioequivalence Assessment of Tofacitinib Once Daily Modified Release Dosage Form in Pediatric Subjects. AAPS J. 2025, 27, 71. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef]
- Kobeissy, P.H.; Denève-Larrazet, C.; Marvaud, J.C.; Kansau, I. MicroRNA miR-27a-5p Reduces Intestinal Inflammation Induced by Clostridioides difficile Flagella by Regulating the Nuclear Factor-κB Signaling Pathway. J. Infect. Dis. 2025, 231, e38–e46. [Google Scholar] [CrossRef]
- Buchner, A.M.; Farraye, F.A.; Iacucci, M. AGA Clinical Practice Update on Endoscopic Scoring Systems in Inflammatory Bowel Disease: Commentary. Clin. Gastroenterol. Hepatol. 2024, 22, 2188–2196. [Google Scholar] [CrossRef]
- Tian, J.; Wang, W.; Liu, Y.; Zhang, X.; Zhao, H.; Qu, H. Role of endoscopic ultrasound as a predictor of histological healing in ulcerative colitis. Ann. Med. 2025, 57, 2499961. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, L.; Hu, T.; Yin, J.; Xu, L.; Pang, Z.; Chen, W. CircRNA_103765 acts as a proinflammatory factor via sponging miR-30 family in Crohn’s disease. Sci. Rep. 2021, 11, 565. [Google Scholar] [CrossRef]
- Nie, J.; Zhao, Q. Lnc-ITSN1-2, Derived From RNA Sequencing, Correlates with Increased Disease Risk, Activity and Promotes CD4(+) T Cell Activation, Proliferation and Th1/Th17 Cell Differentiation by Serving as a ceRNA for IL-23R via Sponging miR-125a in Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 852. [Google Scholar] [CrossRef]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2024, 25, 211–232. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.; Salameh, R.; Karam, K.; Khoury, C.; Fiani, E. A Not-So-Sweet Crohn’s Disease: A Case Report of Ileocecal Crohn’s Disease Unmasked by Sweet Syndrome. Case Rep. Med. 2025, 2025, 6680526. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Li, Y.; Yao, R.; Jiang, M. Osteopontin in Chronic Inflammatory Diseases: Mechanisms, Biomarker Potential, and Therapeutic Strategies. Biology 2025, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- S, D.S.N.; Sundararajan, V. Gene expression analysis reveals mir-29 as a linker regulatory molecule among rheumatoid arthritis, inflammatory bowel disease, and dementia: Insights from systems biology approach. PLoS ONE 2025, 20, e0316584. [Google Scholar] [CrossRef]
- Gole, B.; Potočnik, U. Pre-Treatment Biomarkers of Anti-Tumour Necrosis Factor Therapy Response in Crohn’s Disease—A Systematic Review and Gene Ontology Analysis. Cells 2019, 8, 515. [Google Scholar] [CrossRef]
- Gorenjak, M.; Zupin, M.; Jezernik, G.; Skok, P.; Potočnik, U. Omics data integration identifies ELOVL7 and MMD gene regions as novel loci for adalimumab response in patients with Crohn’s disease. Sci. Rep. 2021, 11, 5449. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene Symbol | Full Gene Name | Tissue | TOFA Regulation | Reference |
|---|---|---|---|---|
| Il1b (mouse) | Interleukin 1 Beta | naive murine CD4(+) T cells | Down | [82] |
| Il18 (mouse) | Interleukin 18 | naive murine CD4(+) T cells | Down | [82] |
| Tbx21 (mouse) | T-Box Transcription Factor 21 | naive murine CD4(+) T cells | Down | [82] |
| Rorc (mouse) | RAR-Related Orphan Receptor gamma | naive murine CD4(+) T cells | Down | [82] |
| Il23r (mouse) | Interleukin 23 Receptor | naive murine CD4(+) T cells | Down | [82] |
| Il21 (mouse) | Interleukin 21 | naive murine CD4(+) T cells | Down | [82] |
| Ahr (mouse) | Aryl Hydrocarbon Receptor | naive murine CD4(+) T cells | Down | [82] |
| TBX21 | T-Box Transcription Factor 21 | peripheral T cells | Down | [83] |
| ISG15 | ISG15 Ubiquitin-Like Modifier | intestinal macrophages | Down | [61] |
| GBP1 | Guanylate Binding Protein 1 | intestinal macrophages | Down | [61] |
| S100A9 | S100 Calcium-Binding Protein A9 | intestinal macrophages | Down | [61] |
| IFITM3 | Interferon-Induced Transmembrane Protein 3 | intestinal macrophages | Down | [61] |
| AHR | Aryl Hydrocarbon Receptor | intestinal macrophages | Up | [61] |
| IGF1 | Insulin-Like Growth Factor 1 | intestinal macrophages | Up | [61] |
| MAF | MAF BZIP Transcription Factor | intestinal macrophages | Up | [61] |
| IL10RA | Interleukin 10 Receptor Subunit Alpha | intestinal macrophages | Up | [61] |
| Alpi (mouse) | Alkaline Phosphatase, Intestinal | enteroid cultures from murine crypts | Up | [89] |
| Cldn3 (mouse) | Claudin 3 | enteroid cultures from murine crypts | Up | [89] |
| Reg3g (mouse) | Regenerating Islet-Derived 3 Gamma | enteroid cultures from murine crypts | Up | [89] |
| Reg3b (mouse) | Regenerating Islet-Derived 3 Beta | enteroid cultures from murine crypts | Up | [89] |
| CLDN8 | Claudin 8 | rectal mucosa | Up | [95] |
| REG1A | Regenerating Family Member 1 Alpha | rectal mucosa | Down | [95] |
| REG1B | Regenerating Family Member 1 Beta | rectal mucosa | Down | [95] |
| REG3A | Regenerating Family Member 3 Alpha | rectal mucosa | Down | [95] |
| CEACAM3 | CEA Cell Adhesion Molecule 3 | colonic biopsies | Down | [103] |
| CEACAM5 | CEA Cell Adhesion Molecule 5 | colonic biopsies | Down | [103] |
| CEACAM6 | CEA Cell Adhesion Molecule 6 | colonic biopsies | Down | [103] |
| FGFR2 | Fibroblast Growth Factor Receptor 2 | peripheral blood | Down | [71] |
| LRPAP1 | LDL Receptor-Related Protein Associated Protein 1 | peripheral blood | Down | [71] |
| OR2L13 | Olfactory Receptor Family 2 Subfamily L Member 13 | peripheral blood | Up | [71] |
| SLC47A1 | Solute Carrier Family 47 member 1 | colonic biopsies | Up | [90] |
| Cell Type | Regulation | R/NR | References |
|---|---|---|---|
| Inflammatory macrophages | Down | R | [139] |
| Plasma cells | Down | R | [61,139] |
| Neutrophils | Down | R | [139] |
| Inflammatory fibroblasts | Down | R | [139] |
| Epithelial cells | Up | R | [61,139] |
| Stromal cells | Up | R | [61,139] |
| Myeloid cells | Up | NR | [61,139] |
| Inflammatory macrophages | Up | NR | [61,139] |
| Inflammatory fibroblasts | Up | NR | [61] |
| B-lymphocytes | Down | R | [61] |
| Granulocytes | Down | R | [61] |
| Gene Symbol | UC Naïve Regulation | TOFA Regulation | Type of TOFA Biomarker | References |
|---|---|---|---|---|
| CLDN8 | Down | Up | T | [80,95] |
| GUCA2A | Down | Down | T | [80,95] |
| HAVCR1 | Down | Down | T | [80,95] |
| HMGCS2 | Down | Up | T | [80,95] |
| PCK1 | Down | Down | T | [80,95] |
| SLC6A19 | Down | Up | T | [80,95] |
| CHI3L2 | Up | Down | T | [61,80] |
| CXCL5 | Up | Down | T, P | [80,95,129] |
| CXCL6 | Up | Up | P | [80,129] |
| IL17A | Up | Down | P | [80,134] |
| IL1B | Up | Up/Down | T, P | [61,80,81,123] |
| REG1B | Up | Down | T | [80,95] |
| REG3A | Up | Down | T | [80,95] |
| S100A9 | Up | Down | T | [61,80] |
| SAA2 | Up | Down | T | [80,95] |
| SLC26A4 | Up | Down | T | [80,95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bizjak, A.; Gole, B.; Jezernik, G.; Potočnik, U.; Gorenjak, M. Toward Precision Medicine: Molecular Biomarkers of Response to Tofacitinib in Inflammatory Bowel Disease. Genes 2025, 16, 908. https://doi.org/10.3390/genes16080908
Bizjak A, Gole B, Jezernik G, Potočnik U, Gorenjak M. Toward Precision Medicine: Molecular Biomarkers of Response to Tofacitinib in Inflammatory Bowel Disease. Genes. 2025; 16(8):908. https://doi.org/10.3390/genes16080908
Chicago/Turabian StyleBizjak, Anja, Boris Gole, Gregor Jezernik, Uroš Potočnik, and Mario Gorenjak. 2025. "Toward Precision Medicine: Molecular Biomarkers of Response to Tofacitinib in Inflammatory Bowel Disease" Genes 16, no. 8: 908. https://doi.org/10.3390/genes16080908
APA StyleBizjak, A., Gole, B., Jezernik, G., Potočnik, U., & Gorenjak, M. (2025). Toward Precision Medicine: Molecular Biomarkers of Response to Tofacitinib in Inflammatory Bowel Disease. Genes, 16(8), 908. https://doi.org/10.3390/genes16080908