
Structural Variants: Mechanisms, Mapping, and Interpretation in Human Genetics


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction to Structural Variations
2. Clinical Relevance
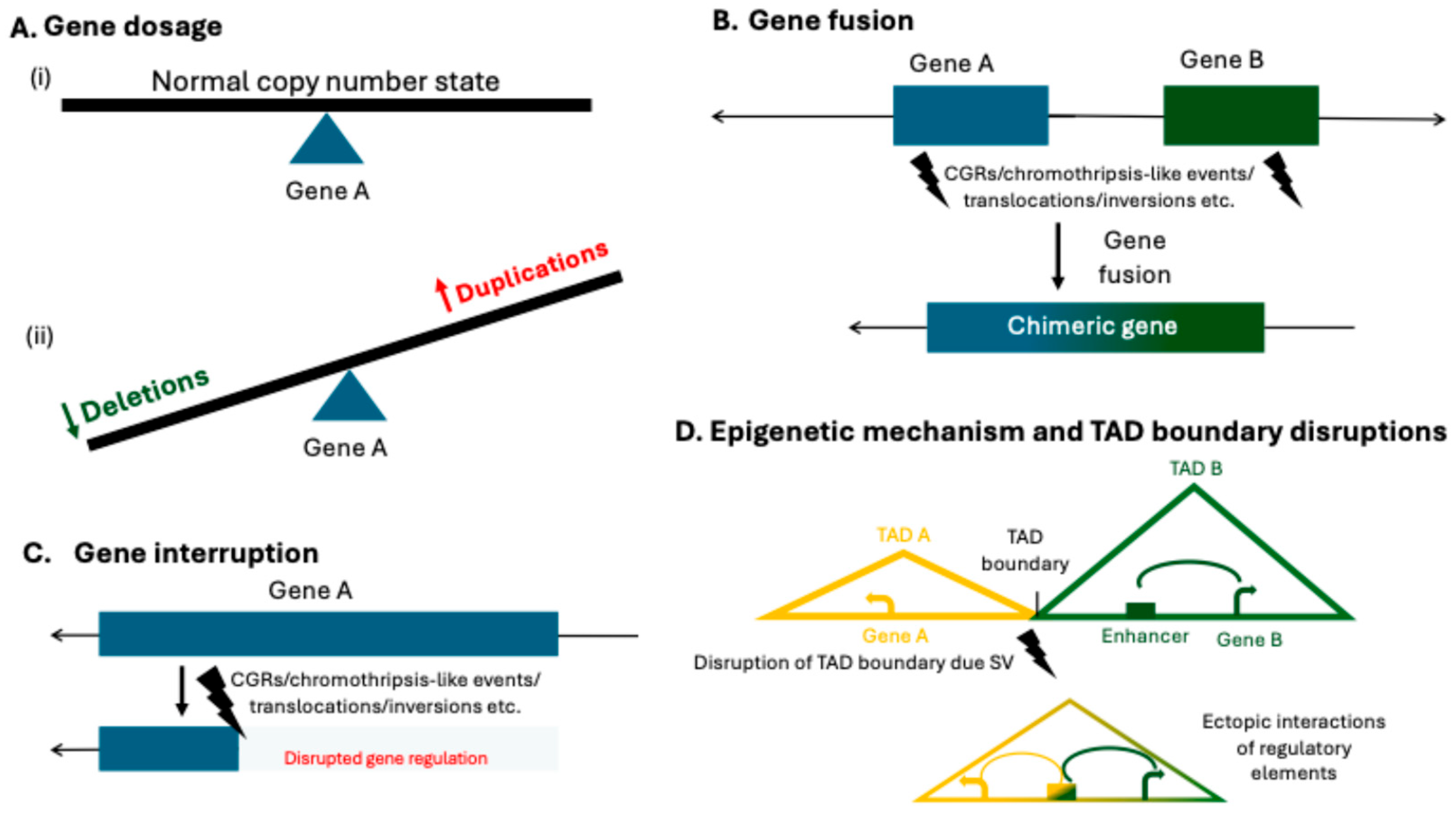
3. Interpretation of Structural Variants
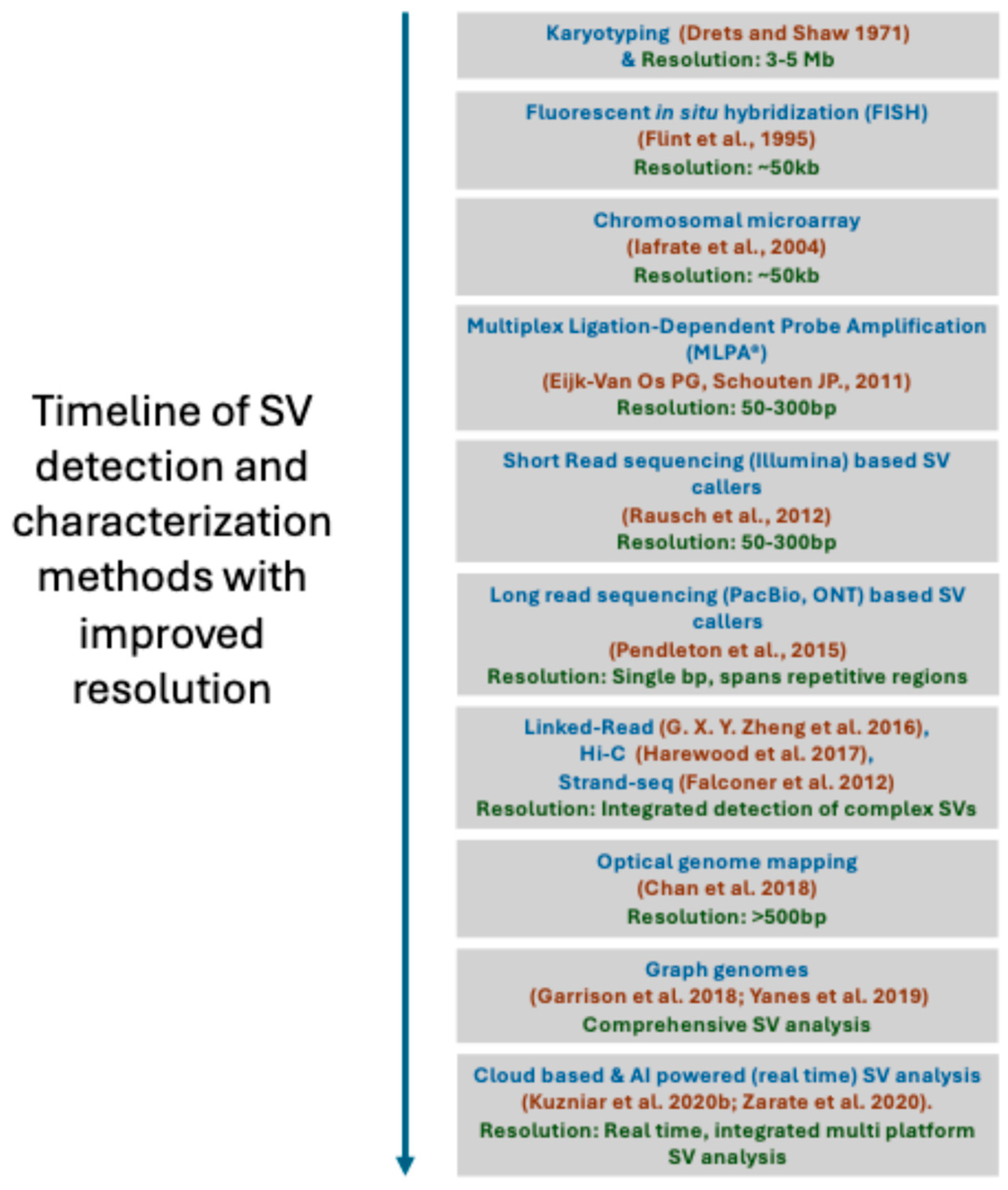
4. Methods for Structural Variant Detection
4.1. Karyotyping
4.2. Chromosomal Microarray
4.3. Targeted CNV Detection
4.4. Optical Genome Mapping
4.5. Structrual Variant Calling Using Next-Generation Sequencing Methods
4.6. SV Callers from Short-Read Whole-Genome Sequencing
4.7. SV Callers from Long-Read Whole-Genome Sequencing
4.8. Strand-Seq
4.9. High-Throughput Chromosome Conformation Capture (Hi-C)
4.10. Linked-Read Sequencing
5. Challenges of Structural Variant Detection, Analysis, and Interpretation
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Rozanski, A.N.; Ryabov, F.; Potapova, T.; Shepelev, V.A.; Catacchio, C.R.; Porubsky, D.; Mao, Y.; Yoo, D.; Rautiainen, M.; et al. The variation and evolution of complete human centromeres. Nature 2024, 629, 136–145. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Stankiewicz, P. Genomic Disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. PLoS Genet. 2005, 1, e49. [Google Scholar] [CrossRef]
- CGrochowski, C.M.; Bengtsson, J.D.; Du, H.; Gandhi, M.; Lun, M.Y.; Mehaffey, M.G.; Park, K.; Höps, W.; Benito, E.; Hasenfeld, P.; et al. Inverted triplications formed by iterative template switches generate structural variant diversity at genomic disorder loci. Cell Genom. 2024, 4, 100590. [Google Scholar] [CrossRef]
- Dardas, Z.; Marafi, D.; Duan, R.; Fatih, J.M.; El-Rashidy, O.F.; Grochowski, C.M.; Carvalho, C.M.; Jhangiani, S.N.; Bi, W.; Du, H.; et al. Genomic Balancing Act: Deciphering DNA rearrangements in the complex chromosomal aberration involving 5p15.2, 2q31.1, and 18q21.32. Eur. J. Hum. Genet. 2024, 33, 231–238. [Google Scholar] [CrossRef]
- Schuy, J.; Grochowski, C.M.; Carvalho, C.M.; Lindstrand, A. Complex genomic rearrangements: An underestimated cause of rare diseases. Trends Genet. 2022, 38, 1134–1146. [Google Scholar] [CrossRef]
- Grochowski, C.M.; Krepischi, A.C.; Eisfeldt, J.; Du, H.; Bertola, D.R.; Oliveira, D.; Costa, S.S.; Lupski, J.R.; Lindstrand, A.; Carvalho, C.M. Chromoanagenesis Event Underlies a de novo Pericentric and Multiple Paracentric Inversions in a Single Chromosome Causing Coffin–Siris Syndrome. Front. Genet. 2021, 12, 708348. [Google Scholar] [CrossRef]
- Pettersson, M.; Grochowski, C.M.; Wincent, J.; Eisfeldt, J.; Breman, A.M.; Cheung, S.W.; Krepischi, A.C.; Rosenberg, C.; Lupski, J.R.; Ottosson, J.; et al. Cytogenetically visible inversions are formed by multiple molecular mechanisms. Hum. Mutat. 2020, 41, 1979–1998. [Google Scholar] [CrossRef]
- Dumas, L.; Kim, Y.H.; Karimpour-Fard, A.; Cox, M.; Hopkins, J.; Pollack, J.R.; Sikela, J.M. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007, 17, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhang, F.; Lupski, J.R. Mechanisms for human genomic rearrangements. Pathogenetics 2008, 1, 4. [Google Scholar] [CrossRef]
- Lupski, J.R. Structural Variation Mutagenesis of the Human Genome: Impact on Disease and Evolution; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Potocki, L.; Bi, W.; Treadwell-Deering, D.; Carvalho, C.M.; Eifert, A.; Friedman, E.M.; Glaze, D.; Krull, K.; Lee, J.A.; Lewis, R.A.; et al. Characterization of Potocki-Lupski Syndrome (dup(17)(p11.2p11.2)) and Delineation of a Dosage-Sensitive Critical Interval That Can Convey an Autism Phenotype. Am. J. Hum. Genet. 2007, 80, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, F.; Lewis, R.A.; Potocki, L.; Glaze, D.; Parke, J.; Killian, J.; Murphy, M.A.; Williamson, D.; Brown, F.; Dutton, R.; et al. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2). Am. J. Med. Genet. 1996, 62, 247–254. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Hastings, P.J.; Ira, G.; Lupski, J.R.; Matic, I. A Microhomology-Mediated Break-Induced Replication Model for the Origin of Human Copy Number Variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Zhang, F.; Khajavi, M.; Connolly, A.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar] [CrossRef]
- Pellestor, F.; Gaillard, J.; Schneider, A.; Puechberty, J.; Gatinois, V. Chromoanagenesis, the mechanisms of a genomic chaos. Semin. Cell Dev. Biol. 2022, 123, 90–99. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Simovic, M.; Ernst, A. Chromothripsis, DNA repair and checkpoints defects. Semin. Cell Dev. Biol. 2022, 123, 110–114. [Google Scholar] [CrossRef]
- Shen, M.M. Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell 2013, 23, 567–569. [Google Scholar] [CrossRef]
- Zepeda-Mendoza, C.J.; Morton, C.C. The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am. J. Hum. Genet. 2019, 104, 565–577. [Google Scholar] [CrossRef]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genom. 2014, 15, 82. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- de Pagter, M.S.; van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; van der Veken, L.T.; Cuppen, E.; et al. Chromothripsis in Healthy Individuals Affects Multiple Protein-Coding Genes and Can Result in Severe Congenital Abnormalities in Offspring. Am. J. Hum. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef]
- Willis, N.A.; Rass, E.; Scully, R. Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer 2015, 1, 217–230. [Google Scholar] [CrossRef]
- Liu, P.; Lacaria, M.; Zhang, F.; Withers, M.; Hastings, P.J.; Lupski, J.R. Frequency of Nonallelic Homol-ogous Recombination Is Correlated with Length of Homology: Evidence that Ectopic Synapsis Precedes Ec-topic Crossing-Over. Am. J. Hum. Genet. 2011, 89, 580–588. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, I.; Lopez, J.; Skipper, L.; Hefferon, T.; Spalding, J.D.; Garner, J.; Chen, C.; Maguire, M.; Corbett, M.; Zhou, G.; et al. dbVar and DGVa: Public archives for genomic structural variation. Nucleic Acids Res. 2012, 41, D936–D941. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef]
- Nicholas, T.J.; Cormier, M.J.; Quinlan, A.R. Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets using SVAFotate. BMC Bioinform. 2022, 23, 490. [Google Scholar] [CrossRef]
- Collins, R.L.; Talkowski, M.E. Diversity and consequences of structural variation in the human genome. Nat. Rev. Genet. 2025, 26, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Laé, M.; Fréneaux, P.; Sastre-Garau, X.; Chouchane, O.; Sigal-Zafrani, B.; Vincent-Salomon, A. Secretory breast carcinomas with ETV6-NTRK3 fusion gene belong to the basal-like carcinoma spectrum. Mod. Pathol. 2009, 22, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, S.; Yun, H.; Cho, S.I.; Kim, B.; Lee, J.-S.; Chae, J.H.; Sun, C.; Park, S.S.; Seong, M.-W. Consistent count region–copy number variation (CCR-CNV): An expandable and robust tool for clinical diagnosis of copy number variation at the exon level using next-generation sequencing data. Genet. Med. 2022, 24, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H. Copy Number Variation Disorders. Curr. Genet. Med. Rep. 2017, 5, 183–190. [Google Scholar] [CrossRef]
- Yuan, B.; Wang, L.; Liu, P.; Shaw, C.; Dai, H.; Cooper, L.; Zhu, W.; Anderson, S.A.; Meng, L.; Wang, X.; et al. CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genet. Med. 2020, 22, 1633–1641. [Google Scholar] [CrossRef]
- da Costa-Nunes, J.A.; Noordermeer, D. TADs: Dynamic structures to create stable regulatory functions. Curr. Opin. Struct. Biol. 2023, 81, 102622. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Daly, A.F.; Dunnington, L.A.; Rodriguez-Buritica, D.F.; Spiegel, E.; Brancati, F.; Mantovani, G.; Rawal, V.M.; Faucz, F.R.; Hijazi, H.; Caberg, J.-H.; et al. Chromatin conformation capture in the clinic: 4C-seq/HiC distinguishes pathogenic from neutral duplications at the GPR101 locus. Genome Med. 2024, 16, 112. [Google Scholar] [CrossRef]
- Cova, G.; Glaser, J.; Schöpflin, R.; Prada-Medina, C.A.; Ali, S.; Franke, M.; Falcone, R.; Federer, M.; Ponzi, E.; Ficarella, R.; et al. Combinatorial effects on gene expression at the Lbx1/Fgf8 locus resolve split-hand/foot malformation type 3. Nat. Commun. 2023, 14, 1475. [Google Scholar] [CrossRef]
- Salnikov, P.; Korablev, A.; Serova, I.; Belokopytova, P.; Yan, A.; Stepanchuk, Y.; Tikhomirov, S.; Fishman, V. Structural variants in the Epb41l4a locus: TAD disruption and Nrep gene misregulation as hypothetical drivers of neurodevelopmental outcomes. Sci. Rep. 2024, 14, 5288. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Tjio, J.H.; Levan, A. The Chromosome Number Of Man. Hereditas 2010, 42, 1–6. [Google Scholar] [CrossRef]
- Harel, T.; Lupski, J. Genomic Disorders 20 Years on—Mechanisms for Clinical Manifestations; Blackwell Publishing Ltd.: Oxford, UK, 2018. [Google Scholar] [CrossRef]
- Drets, M.E.; Shaw, M.W. Specific Banding Patterns of Human Chromosomes (heterochromatin/Giemsa stain/chromosome bands). Proc. Natl. Acad. Sci. USA 1971, 68, 2073–2077. [Google Scholar] [CrossRef]
- A Ferguson-Smith, M. History and evolution of cytogenetics. Mol. Cytogenet. 2015, 8, 19. [Google Scholar] [CrossRef]
- Teixeira, W.G.; Marques, F.K.; Freire, M.C.M. Retrospective Karyotype Study in Mentally Retarded Patients. Rev. Assoc. Med. Bras. 2016, 62, 262–268. [Google Scholar] [CrossRef]
- Grochowski, C.M.; Gu, S.; Yuan, B.; Tcw, J.; Brennand, K.J.; Sebat, J.; Malhotra, D.; McCarthy, S.; Rudolph, U.; Lindstrand, A.; et al. Marker Chromosome Genomic Structure and Temporal Origin Implicate a Chro-moanasynthesis Event in a Family with Pleiotropic Psychiatric Phenotypes. Hum. Mutat. 2018, 39, 939–946. [Google Scholar] [CrossRef]
- Hollenbeck, D.; Williams, C.L.; Drazba, K.; Descartes, M.; Korf, B.R.; Rutledge, S.L.; Lose, E.J.; Robin, N.H.; Carroll, A.J.; Mikhail, F.M. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet. Med. 2017, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Lee, J.-Y.; Blake, D.M.; Bunke, B.P.; Alexander, C.R.; Kogan, A.L.; Ledbetter, D.H.; Martin, C.L. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet. Med. 2008, 10, 415–429. [Google Scholar] [CrossRef]
- Gijsbers, A.; Schoumans, J.; Ruivenkamp, C. Interpretation of Array Comparative Genome Hybridization Data: A Major Challenge. Cytogenet. Genome Res. 2011, 135, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Cai, L.; Yao, H.; Dong, X. Chromosome microarray analysis combined with karyotype analysis is a powerful tool for the detection in pregnant women with high-risk indicators. BMC Pregnancy Childbirth 2023, 23, 784. [Google Scholar] [CrossRef] [PubMed]
- Flint, J.; Wilkie, A.O.; Buckle, V.J.; Winter, R.M.; Holland, A.J.; McDermid, H.E. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat. Genet. 1995, 9, 132–140. [Google Scholar] [CrossRef]
- Eijk-Van Os, P.G.C.; Schouten, J.P. Multiplex Ligation-Dependent Probe Amplification (MLPA®) for the Detection of Copy Number Variation in Genomic Sequences; Springer Nature: Berlin/Heidelberg, Germany, 2011; pp. 97–126. [Google Scholar] [CrossRef]
- Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA Assay in the Molecular Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases. Int. J. Mol. Sci. 2012, 13, 3245–3276. [Google Scholar] [CrossRef]
- Chan, E.K.; Cameron, D.L.; Petersen, D.C.; Lyons, R.J.; Baldi, B.F.; Papenfuss, A.T.; Thomas, D.M.; Hayes, V.M. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res. 2018, 28, 726–738. [Google Scholar] [CrossRef]
- Talenti, A.; Powell, J.; Wragg, D.; Chepkwony, M.; Fisch, A.; Ferreira, B.R.; Mercadante, M.E.Z.; Santos, I.M.; Ezeasor, C.K.; Obishakin, E.T.; et al. Optical mapping compendium of structural variants across global cattle breeds. Sci. Data 2022, 9, 618. [Google Scholar] [CrossRef]
- Schrauwen, I.; Rajendran, Y.; Acharya, A.; Öhman, S.; Arvio, M.; Paetau, R.; Siren, A.; Avela, K.; Granvik, J.; Leal, S.M.; et al. Optical genome mapping unveils hidden structural variants in neurodevelopmental disorders. Sci. Rep. 2024, 14, 11239. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, D.; Bengtsson, J.D.; Bajikar, S.S.; Grochowski, C.M.; Lun, M.Y.; Gandhi, M.; Jolly, A.; Trostle, A.J.; Harris, H.K.; Suter, B.; et al. Structural variant allelic heterogeneity in MECP2 duplication syndrome provides insight into clinical severity and variability of disease expression. Genome Med. 2024, 16, 146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bai, W.; Yuan, N.; Du, Z.; Ioshikhes, I. Comprehensively benchmarking applications for detecting copy number variation. PLoS Comput. Biol. 2019, 15, e1007069. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Kutzner, A. MSV: A modular structural variant caller that reveals nested and complex rearrangements by unifying breakends inferred directly from reads. Genome Biol. 2023, 24, 170. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- English, A.C.; Salerno, W.J.; A Hampton, O.; Gonzaga-Jauregui, C.; Ambreth, S.; I Ritter, D.; Beck, C.R.; Davis, C.F.; Dahdouli, M.; Ma, S.; et al. Assessing structural variation in a personal genome—Towards a human reference diploid genome. BMC Genom. 2015, 16, 286. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Yi, D.; Nam, J.-W.; Jeong, H. Toward the functional interpretation of somatic structural variations: Bulk-and single-cell approaches. Brief. Bioinform. 2023, 24, bbad297. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 1865–2871. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Kuzniar, A.; Maassen, J.; Verhoeven, S.; Santuari, L.; Shneider, C.; Kloosterman, W.P.; de Ridder, J. sv-callers: A highly portable parallel workflow for structural variant detection in whole-genome sequence data. PeerJ 2020, 8, e8214. [Google Scholar] [CrossRef]
- Cameron, D.L.; Schröder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P.; Papenfuss, A.T. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Li, T.; Vermeesch, J.R.; Xie, Z. A benchmark of structural variation detection by long reads through a realistic simulated model. Genome Biol. 2021, 22, 342. [Google Scholar] [CrossRef]
- Smith, S.D.; Kawash, J.K.; Grigoriev, A. Lightning-fast genome variant detection with GROM. Gigascience 2017, 6, gix091. [Google Scholar] [CrossRef]
- English, A.C.; Salerno, W.J.; Reid, J.G. PBHoney: Identifying Genomic Variants via Long-read Discord-ance and Interrupted mapping. BMC Bioinform. 2014, 15, 180. [Google Scholar] [CrossRef]
- Stancu, M.C.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-Inclan, J.E.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [PubMed]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360, eaar6343. [Google Scholar] [CrossRef]
- Smolka, M.; Paulin, L.F.; Grochowski, C.M.; Horner, D.W.; Mahmoud, M.; Behera, S.; Kalef-Ezra, E.; Gandhi, M.; Hong, K.; Pehlivan, D.; et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. 2024, 42, 1571–1580. [Google Scholar] [CrossRef]
- Heller, D.; Vingron, M. SVIM: Structural variant identification using mapped long reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, T.; Su, J.; Liu, B.; Zang, T.; Wang, Y.; Marschall, T. SKSV: Ultrafast structural variation detection from circular consensus sequencing reads. Bioinformatics 2021, 37, 3647–3649. [Google Scholar] [CrossRef]
- Ding, H.; Luo, J. MAMnet: Detecting and genotyping deletions and insertions based on long reads and a deep learning approach. Brief. Bioinform. 2022, 23, bbac195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, A.Y.; Barkley, C.A.; Zhang, Y.; Zhao, X.; Gao, M.; Edmonds, M.D.; Chong, Z. Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak. Nat. Commun. 2023, 14, 283. [Google Scholar] [CrossRef]
- Earl, D.; Bradnam, K.; John, J.S.; Darling, A.; Lin, D.; Fass, J.; Yu, H.O.K.; Buffalo, V.; Zerbino, D.R.; Diekhans, M.; et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21, 2224–2241. [Google Scholar] [CrossRef]
- Lee, H.; Gurtowski, J.; Yoo, S.; Marcus, S.; McCombie, W.R.; Schatz, M. Error correction and assembly complexity of single molecule sequencing reads. bioRxiv 2014. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Luo, R.; Wu, H.; Zhu, H.; Li, R.; Cao, H.; Wu, B.; Huang, S.; Shao, H.; et al. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat. Biotechnol. 2011, 29, 723–730. [Google Scholar] [CrossRef]
- Chong, Z.; Ruan, J.; Gao, M.; Zhou, W.; Chen, T.; Fan, X.; Ding, L.; Lee, A.Y.; Boutros, P.; Chen, J.; et al. novoBreak: Local assembly for breakpoint detection in cancer genomes. Nat. Methods 2017, 14, 65–67. [Google Scholar] [CrossRef]
- Nattestad, M.; Schatz, M.C. Assemblytics: A web analytics tool for the detection of variants from an assembly. Bioinformatics 2016, 32, 3021–3023. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A.; Darling, A.E. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.L.; Di Stefano, L.; Papenfuss, A.T. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat. Commun. 2019, 10, 3240. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Lun, M.Y.; Gagarina, L.; Mehaffey, M.G.; Hwang, J.P.; Jhangiani, S.N.; Bhamidipati, S.V.; Muzny, D.M.; Poli, M.C.; Ochoa, S.; et al. VizCNV: An integrated platform for concurrent phased BAF and CNV analysis with trio genome sequencing data. bioRxiv 2024. preprint. [Google Scholar] [CrossRef] [PubMed]
- Wala, J.A.; Bandopadhayay, P.; Greenwald, N.F.; O’ROurke, R.; Sharpe, T.; Stewart, C.; Schumacher, S.; Li, Y.; Weischenfeldt, J.; Yao, X.; et al. SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018, 28, 581–591. [Google Scholar] [CrossRef]
- Chen, S.; Krusche, P.; Dolzhenko, E.; Sherman, R.M.; Petrovski, R.; Schlesinger, F.; Kirsche, M.; Bentley, D.R.; Schatz, M.C.; Sedlazeck, F.J.; et al. Paragraph: A graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019, 20, 291. [Google Scholar] [CrossRef]
- Mohiyuddin, M.; Mu, J.C.; Li, J.; Asadi, N.B.; Gerstein, M.B.; Abyzov, A.; Wong, W.H.; Lam, H.Y. MetaSV: An accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics 2015, 31, 2741–2744. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Mahmoud, M.; Agustinho, D.P.; Sedlazeck, F.J. A Hitchhiker’s Guide to long-read genomic analysis. Genome Res. 2025, 35, 545–558. [Google Scholar] [CrossRef]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Mari, R.S.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117. [Google Scholar] [CrossRef]
- Genomics, N.C.F.C.D.; Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Sulovari, A.; Li, R.; Audano, P.A.; Porubsky, D.; Vollger, M.R.; Logsdon, G.A.; Warren, W.C.; Pollen, A.A.; Chaisson, M.J.P.; Eichler, E.E. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. USA 2019, 116, 23243–23253. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Liu, Q.; Perdomo, J.E.; Fang, L.; Wang, K. A survey of algorithms for the detection of genomic structural variants from long-read sequencing data. Nat. Methods 2023, 20, 1143–1158. [Google Scholar] [CrossRef]
- Aydin, S.K.; Yilmaz, K.C.; Acar, A. Benchmarking long-read structural variant calling tools and combinations for detecting somatic variants in cancer genomes. Sci. Rep. 2025, 15, 8707. [Google Scholar] [CrossRef] [PubMed]
- Helal, A.A.; Saad, B.T.; Saad, M.T.; Mosaad, G.S.; Aboshanab, K.M. Benchmarking long-read aligners and SV callers for structural variation detection in Oxford nanopore sequencing data. Sci. Rep. 2024, 14, 6160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Denti, L.; Khorsand, P.; Bonizzoni, P.; Hormozdiari, F.; Chikhi, R. SVDSS: Structural variation discovery in hard-to-call genomic regions using sample-specific strings from accurate long reads. Nat. Methods 2023, 20, 550–558. [Google Scholar] [CrossRef]
- Zheng, Y.; Shang, X.; Sung, W.-K. SVsearcher: A more accurate structural variation detection method in long read data. Comput. Biol. Med. 2023, 158, 106843. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shang, X. SVvalidation: A long-read-based validation method for genomic structural varia-tion. PLoS ONE 2024, 19, e0291741. [Google Scholar] [CrossRef]
- Zheng, Y.; Shang, X. SVcnn: An accurate deep learning-based method for detecting structural variation based on long-read data. BMC Bioinform. 2023, 24, 213. [Google Scholar] [CrossRef]
- Xia, Z.; Xiang, W.; Wang, Q.; Li, X.; Li, Y.; Gao, J.; Tang, T.; Yang, C.; Cui, Y.; Birol, I. CSV-Filter: A deep learning-based comprehensive structural variant filtering method for both short and long reads. Bioinformatics 2024, 40, btae539. [Google Scholar] [CrossRef]
- Linderman, M.D.; Wallace, J.; van der Heyde, A.; Wieman, E.; Brey, D.; Shi, Y.; Hansen, P.; Shamsi, Z.; Liu, J.; Gelb, B.D.; et al. NPSV-deep: A deep learning method for genotyping structural variants in short read genome sequencing data. Bioinformatics 2024, 40, btae129. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Sun, J.; Wang, X.; Wang, J.; Liu, Q.; Ru, J.; Zhang, X.; Wang, S.; Hao, R.; Bian, P.; et al. SVLearn: A dual-reference machine learning approach enables accurate cross-species genotyping of structural variants. Nat. Commun. 2025, 16, 2406. [Google Scholar] [CrossRef] [PubMed]
- Niehus, S.; Jónsson, H.; Schönberger, J.; Björnsson, E.; Beyter, D.; Eggertsson, H.P.; Sulem, P.; Stefánsson, K.; Halldórsson, B.V.; Kehr, B. PopDel identifies medium-size deletions simultaneously in tens of thousands of genomes. Nat. Commun. 2021, 12, 730. [Google Scholar] [CrossRef]
- Mirus, T.; Lohmayer, R.; Döhring, C.; Halldórsson, B.V.; Kehr, B. GGTyper: Genotyping complex structural variants using short-read sequencing data. Bioinformatics 2024, 40, ii11–ii19. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H.; et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef]
- Falconer, E.; Hills, M.; Naumann, U.; Poon, S.S.S.; A Chavez, E.; Sanders, A.D.; Zhao, Y.; Hirst, M.; Lansdorp, P.M. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 2012, 9, 1107–1112. [Google Scholar] [CrossRef]
- Claussin, C.; Porubský, D.; Spierings, D.C.; Halsema, N.; Rentas, S.; Guryev, V.; Lansdorp, P.M.; Chang, M. Genome-wide mapping of sister chromatid exchange events in single yeast cells using Strand-seq. Elife 2017, 6, e30560. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.D.; Hills, M.; Porubský, D.; Guryev, V.; Falconer, E.; Lansdorp, P.M. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. 2016, 26, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.D.; Meiers, S.; Ghareghani, M.; Porubsky, D.; Jeong, H.; van Vliet, M.A.C.C.; Rausch, T.; Richter-Pechańska, P.; Kunz, J.B.; Jenni, S.; et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat. Biotechnol. 2020, 38, 343–354. [Google Scholar] [CrossRef]
- Weber, T.; Cosenza, M.R.; Korbel, J. MosaiCatcher v2: A single-cell structural variations detection and analysis reference framework based on Strand-seq. Bioinformatics 2023, 39, btad633. [Google Scholar] [CrossRef]
- Harewood, L.; Kishore, K.; Eldridge, M.D.; Wingett, S.; Pearson, D.; Schoenfelder, S.; Collins, V.P.; Fraser, P. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol. 2017, 18, 125. [Google Scholar] [CrossRef]
- Song, F.; Xu, J.; Dixon, J.; Yue, F. Analysis of Hi-C Data for Discovery of Structural Variations in Cancer. Methods Mol. Biol. 2022, 2301, 143–161. [Google Scholar] [CrossRef]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, L.; Ye, Y. HiSV: A control-free method for structural variation detection from Hi-C data. PLoS Comput. Biol. 2023, 19, e1010760. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Greer, S.U.; Ji, H.P. Structural variant analysis for linked-read sequencing data with gemtools. Bioinformatics 2019, 35, 4397–4399. [Google Scholar] [CrossRef]
- Fang, L.; Kao, C.; Gonzalez, M.V.; Mafra, F.A.; Da Silva, R.P.; Li, M.; Wenzel, S.-S.; Wimmer, K.; Hakonarson, H.; Wang, K. LinkedSV for detection of mosaic structural variants from linked-read exome and genome sequencing data. Nat. Commun. 2019, 10, 5585. [Google Scholar] [CrossRef]
- Gunturkun, M.H.; Villani, F.; Colonna, V.; Ashbrook, D.; Williams, R.W.; Chen, H. SVJAM: Joint Analysis of Structural Variants Using Linked Read Sequencing Data. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Guichard, A.; Legeai, F.; Tagu, D.; Lemaitre, C. MTG-Link: Leveraging barcode information from linked-reads to assemble specific loci. BMC Bioinform. 2023, 24, 284. [Google Scholar] [CrossRef]
- Yanes, L.; Accinelli, G.G.; Wright, J.; Ward, B.J.; Clavijo, B.J. A Sequence Distance Graph framework for genome assembly and analysis. F1000Research 2019, 8, 1490. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural Variant Calling: The Long and the Short of It; BioMed Central Ltd.: London, UK, 2019. [Google Scholar] [CrossRef]
- Bezdvornykh, I.; Cherkasov, N.; Kanapin, A.; Samsonova, A. A collection of read depth profiles at structural variant breakpoints. Sci. Data 2023, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Joe, S.; Park, J.-L.; Kim, J.; Kim, S.; Park, J.-H.; Yeo, M.-K.; Lee, D.; Yang, J.O.; Kim, S.-Y. Comparison of structural variant callers for massive whole-genome sequence data. BMC Genom. 2024, 25, 318. [Google Scholar] [CrossRef] [PubMed]
- Zook, J.M.; Hansen, N.F.; Olson, N.D.; Chapman, L.; Mullikin, J.C.; Xiao, C.; Sherry, S.; Koren, S.; Phillippy, A.M.; Boutros, P.C.; et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 2020, 38, 1347–1355. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Rautiainen, M.; Marschall, T. GraphAligner: Rapid and versatile sequence-to-graph alignment. Genome Biol. 2020, 21, 253. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Saether, K.B.; Bengtsson, J.; Eisfeldt, J.; Lun, M.Y.; Schuy, J.; Mahmoud, M.; Grochowski, C.M.; Pehlivan, D.; Sedlazeck, F.J.; et al. 11: To remap or not to remap: The relevance of the genome references to resolve rare inversions. Genet. Med. Open 2025, 3, 101928. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Saether, K.B.; Bengtsson, J.; Eisfeldt, J.; Lun, M.Y.; Schuy, J.; Mahmoud, M.; Grochowski, C.M.; Pehlivan, D.; Sedlazeck, F.J.; et al. A complete reference genome improves analysis of human genetic variation. Science 2022, 376, eabl3533. [Google Scholar] [CrossRef]
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Chaisson, M.J.; Steinberg, K.M.; Warren, W.; Hoekzema, K.; Gordon, D.; Graves-Lindsay, T.A.; Munson, K.M.; Kronenberg, Z.N.; Vives, L.; et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017, 27, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; O’nEill, H.; Wolvetang, E.J.; Chatterjee, A.; Gupta, I. Advances in single-cell long-read sequencing technologies. NAR Genom. Bioinform. 2024, 6, lqae047. [Google Scholar] [CrossRef]
- Zarate, S.; Carroll, A.; Mahmoud, M.; Krasheninina, O.; Jun, G.; Salerno, W.J.; Schatz, M.C.; Boerwinkle, E.; A Gibbs, R.; Sedlazeck, F.J. Parliament2: Accurate structural variant calling at scale. Gigascience 2020, 9, giaa145. [Google Scholar] [CrossRef]
- Dawood, M.; Heavner, B.; Wheeler, M.M.; Ungar, R.A.; LoTempio, J.; Wiel, L.; Berger, S.; Bernstein, J.A.; Chong, J.X.; Délot, E.C.; et al. GREGoR: Accelerating Genomics for Rare Diseases. arXiv 2024, arXiv:2412.14338. [Google Scholar]
- GTEx Consortium; Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Karageorgiou, C.; Gokcumen, O.; Dennis, M.Y. Deciphering the role of structural variation in human evolution: A functional perspective. Curr. Opin. Genet. Dev. 2024, 88, 102240. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J.; Berger, B. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Ganel, L.; Abel, H.J.; FinMetSeq Consortium; Hall, I.M. SVScore: An impact prediction tool for structural variation. Bioinformatics 2017, 33, 1083–1085. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pande, S.; Dawood, M.; Grochowski, C.M. Structural Variants: Mechanisms, Mapping, and Interpretation in Human Genetics. Genes 2025, 16, 905. https://doi.org/10.3390/genes16080905
Pande S, Dawood M, Grochowski CM. Structural Variants: Mechanisms, Mapping, and Interpretation in Human Genetics. Genes. 2025; 16(8):905. https://doi.org/10.3390/genes16080905
Chicago/Turabian StylePande, Shruti, Moez Dawood, and Christopher M. Grochowski. 2025. "Structural Variants: Mechanisms, Mapping, and Interpretation in Human Genetics" Genes 16, no. 8: 905. https://doi.org/10.3390/genes16080905
APA StylePande, S., Dawood, M., & Grochowski, C. M. (2025). Structural Variants: Mechanisms, Mapping, and Interpretation in Human Genetics. Genes, 16(8), 905. https://doi.org/10.3390/genes16080905