
Gene-by-Environment Interactions Involving Maternal Exposures with Orofacial Cleft Risk in Filipinos

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population Description
2.2. Genotyping and Imputation
2.3. Statistical Analyses
2.3.1. Gene-by-Environment Interaction Analyses
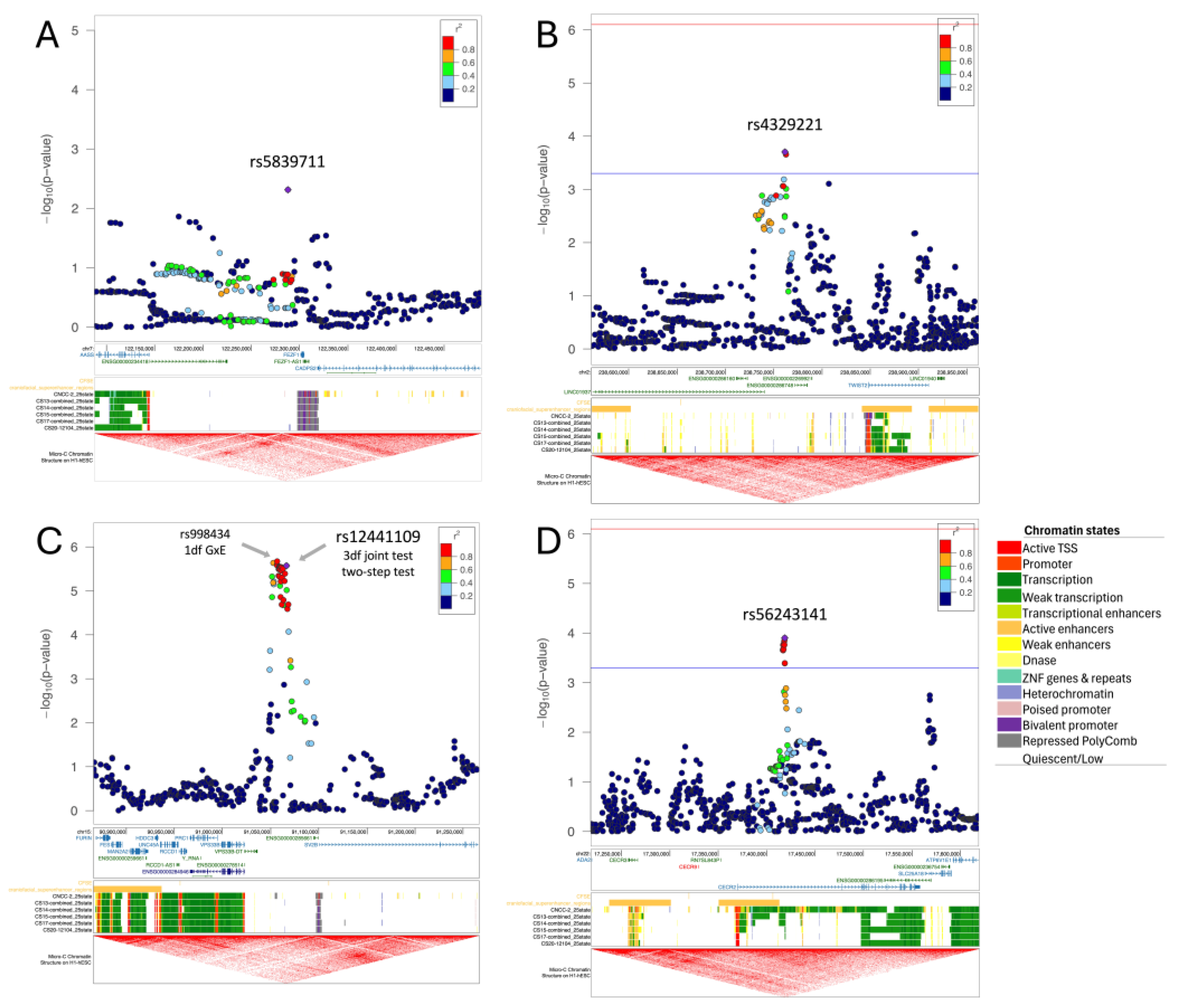
2.3.2. Regional Plots
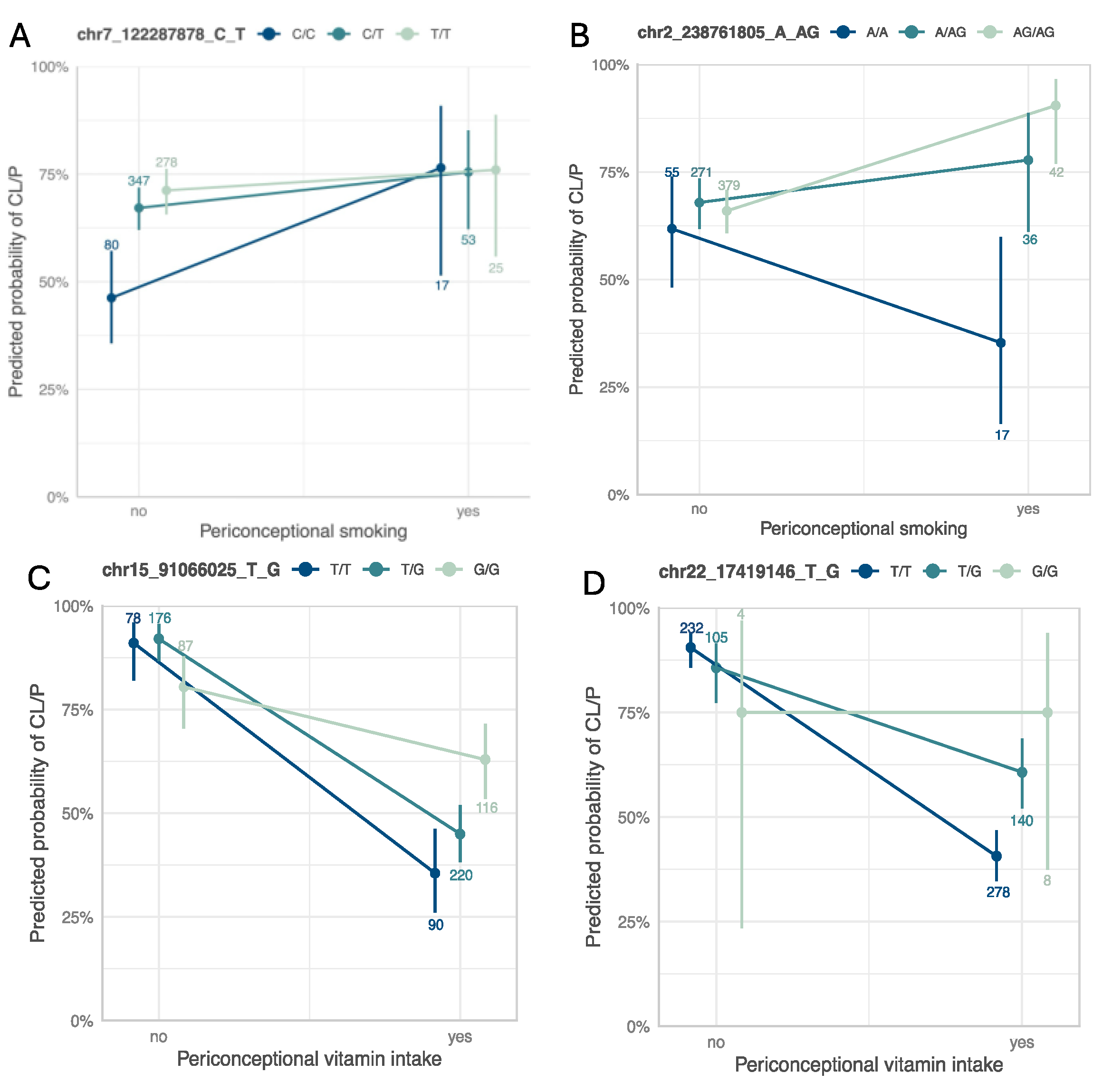
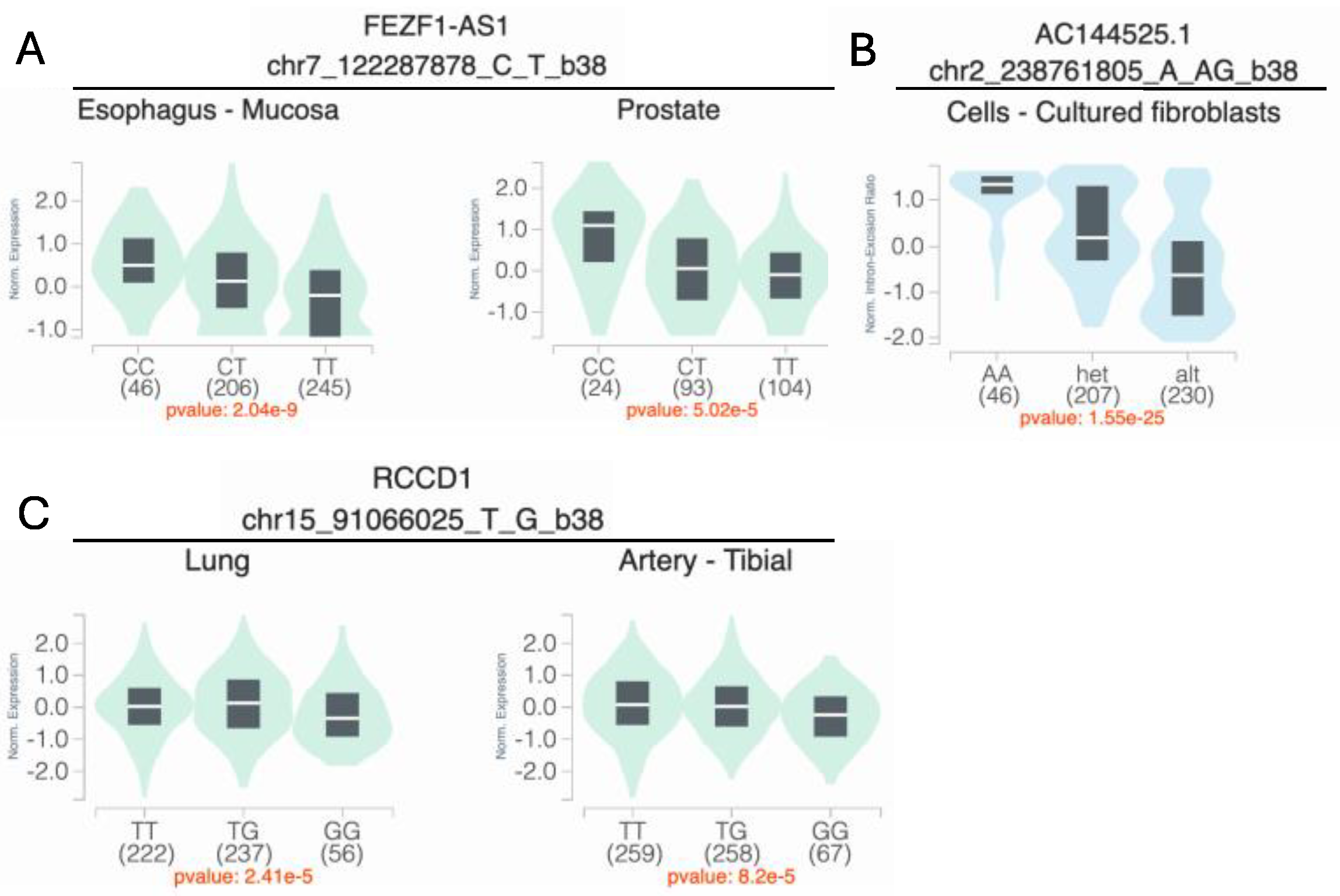
2.3.3. Interaction Plots and eQTL/sQTL Plots
2.3.4. Sensitivity Analysis
2.3.5. Replication
2.3.6. Examination of Known GEI Loci Implicated in CL/P Risk in This GEI Analysis
3. Results
3.1. Genome-Wide Interaction Analysis for Maternal Smoking

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure | Method | Variant Info | Discovery | Replication 1 | Replication 2 | Replication 3 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNV (hg38) | rsID | Nearest Gene | Type | Alt AF | 3df Test | 2df EDGE | GxE | DG | GE All | GE Cases | GE Controls | Case Only | Case Only | Case Only | |||||||
| P | P | β | P | P | P | β | P | P | β | P | β | P | β | P | |||||||
| Smoking | 3df test | 7-122287878-C-T | rs4329221 | FEZF1 | intergenic | 0.63 | 9.87 × 10−6 | 6.18 × 10−6 | −0.58 | 1.63 × 10−1 | 2.87 × 10−5 | 1.09 × 10−2 | −0.57 | 4.87 × 10−3 | 5.71 × 10−1 | −0.06 a | 9.10 × 10−1 | −1.39 a | 2.63 × 10−1 | −0.87 | 3.44 × 10−3 |
| Smoking | Two-step test | 2-238761805-A-AG | rs5839711 | TWIST2 | intergenic | 0.72 | 7.93 × 10−5 | 2.08 × 10−2 | 1.56 | 1.98 × 10−4 | 6.62 × 10−1 | 6.00 × 10−3 | −0.07 | 7.53 × 10−1 | 1.31 × 10−5 a | 0.15 | 8.22 × 10−1 | 50.57 a | 3.50 × 10−1 | 0.61 | 7.15 × 10−2 |
| Vitamin intake | 3df test and two-step test | 15-91066025-T-G | rs12441109 | SV2B | intergenic | 0.53 | 2.65 × 10−6 | 3.66 × 10−7 | 1.45 | 2.68 × 10−6 | 2.02 × 10−1 | 2.55 × 10−2 | 0.65 | 1.02 × 10−5 | 9.69 × 10−3 | −0.25 | 4.51 × 10−1 | 0.04 | 9.34 × 10−1 | 0.03 | 9.02 × 10−1 |
| Vitamin intake | Two-step test | 22-17419146-T-G | rs56243141 | CECR2 | intronic | 0.17 | 2.58 × 10−4 | 2.52 × 10−2 | 1.56 | 1.26 × 10−4 | 2.28 × 10−2 | 1.39 × 10−1 | 0.65 | 4.98 × 10−4 | 2.60 × 10−3 | −0.123 | 7.90 × 10−1 | 0.71 | 2.21 × 10−1 | −0.36 | 3.11 × 10−1 |
| Vitamin intake | 3df test and two-step test | 5-57049741-A-G | rs179464 | MIER3 | intergenic | 0.51 | 8.33 × 10−6 | 1.02 × 10−3 | −1.20 | 4.04 × 10−4 | 2.54 × 10−4 | 5.34 × 10−1 | −0.18 | 2.31 × 10−1 | 4.43 × 10−5 | −0.50 | 1.83 × 10−1 | 0.10 | 8.42 × 10−1 | −0.13 | 7.02 × 10−1 |
3.2. Genome-Wide Interaction Analysis for Maternal Vitamin Use
3.3. Replication Analysis
3.4. Known GEI Loci with Maternal Smoking and Vitamin Intake in CL/P Risk
| Exposure | Variant Info | Discovery Sample | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNV (hg38) | rsID | Nearest Gene | Type | Study MAF | Reference | AF | GxE | DG | GE Cases | ||||
| OR | P | OR | P | OR | P | ||||||||
| Smoking | 4-92455080-G-A | rs4389540 | GRID2 | Intronic | 0.14 | Beaty et al. (2013) [14] | 0.005 | - | - | - | - | - | - |
| Smoking | 7-150999023-T-G | rs1799983 | NOS3 | Exonic-missense | 0.26 | Shaw et al. (2005) [13] | 0.17 | 0.76 | 6.16 × 10−1 | 0.99 | 9.33 × 10−1 | 0.91 | 7.19 × 10−1 |
| Smoking | 9-24527359-G-A | rs2257210 | ELAVL2 | Intergenic | 0.31 | Beaty et al. (2013) [14] | 0.21 | 1.11 | 8.23 × 10−1 | 0.97 | 8.24 × 10−1 | 1.31 | 2.30 × 10−1 |
| Vitamin intake | 1-216999264-T-C | rs1339221 | ESRRG | Intronic | 0.40 | Haaland et al. (2018) [17] | 0.32 | 0.54 | 6.01 × 10−2 | 1.03 | 8.33 × 10−1 | 0.83 | 2.03 × 10−1 |
| Vitamin intake | 7-150999023-T-G | rs1799983 | NOS3 | Exonic-missense | 0.26 | Shaw et al. (2005) [13] | 0.17 | 3.93 | 6.42 × 10−3 | 0.85 | 3.60 × 10−1 | 1.04 | 8.59 × 10−1 |
| Vitamin intake | 16-24342036-A-G | rs9930171 | CACNG3 | Intronic | 0.35 | Carlson et al. (2022) [16] | 0.41 | 0.85 | 6.04 × 10−1 | 0.93 | 5.92 × 10−1 | 0.96 | 7.60 × 10−1 |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CL/P | Cleft lip with or without cleft palate |
| df | Degree-of-freedom |
| DG | Disease–gene association, referring to genetic main effect |
| eQTL | Expression quantitative trait locus |
| GE | Environment–gene association |
| GEI | Gene–environment interaction |
| OFC | Orofacial cleft |
| POFC | Pittsburgh Orofacial Cohort |
| sQTL | Splicing quantitative trait locus |
References
- Murray, J.C.; Daack-Hirsch, S.; Buetow, K.H.; Munger, R.; Espina, L.; Paglinawan, N.; Villanueva, E.; Rary, J.; Magee, K.; Magee, W. Clinical and epidemiologic studies of cleft Lip and palate in the Philippines. Cleft Palate Craniofacial J. 1997, 34, 7–10. [Google Scholar] [CrossRef]
- David-Padilla, C.; Paz EMCC, la.; Lucero, F.; Villafuerte, C.; Cardenas, J.; Villanueva, E. Profile of Oral Cleft Cases Reported in the Philippine Oral Cleft Registry from May 2003 to December 2006. Acta Med. Philipp. 2008, 42, 27–33. [Google Scholar]
- Jugessur, A.; Murray, J.C. Orofacial clefting: Recent insights into a complex trait. Curr. Opin. Genet. Dev. 2005, 15, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Munger, R.G.; Corcoran, C.; Bacayao, J.Y.; Nepomuceno, B.; Solon, F. Plasma zinc concentrations of mothers and the risk of nonsyndromic oral clefts in their children: A case-control study in the Philippines. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Munger, R.G.; Nepomuceno, B.; Corcoran, C.; Cembrano, J.; Solon, F. Maternal plasma pyridoxal-5′-phosphate concentrations and risk of isolated oral clefts in the Philippines. Birth Defects Res. Part A Clin. Mol. Teratol. 2007, 79, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Munger, R.G.; Sauberlich, H.E.; Corcoran, C.; Nepomuceno, B.; Daack-Hirsch, S.; Solon, F.S. Maternal vitamin B-6 and folate status and risk of oral cleft birth defects in the Philippines. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Ly, S.; Magee, K.S.; Ihenacho, U.; Baurley, J.W.; Sanchez-Lara, P.A.; Brindopke, F.; Nguyen, T.; Nguyen, V.; Tangco, M.I.; et al. Parental risk factors for oral clefts among Central Africans, Southeast Asians, and Central Americans. Birth Defects Res. Part A Clin. Mol. Teratol. 2015, 103, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Rajabi, A.; Gholian-Aval, M.; Peyman, N.; Mahdizadeh, M.; Tehrani, H. National, regional, and global prevalence of cigarette smoking among women/females in the general population: A systematic review and meta-analysis. Environ. Health Prev. Med. 2021, 26, 5. [Google Scholar] [CrossRef] [PubMed]
- Auslander, A.; McKean-Cowdin, R.; Brindopke, F.; Sylvester, B.; DiBona, M.; Magee, K.; Kapoor, R.; Conti, D.V.; Rakotoarison, S.; Magee, W. The role of smoke from cooking indoors over an open flame and parental smoking on the risk of cleft lip and palate: A case-control study in 7 low-resource countries. J. Glob. Health 2020, 10, 020410. [Google Scholar] [CrossRef] [PubMed]
- Marazita, M.L. Gene × environment associations in orofacial clefting. Curr. Top. Dev. Biol. 2023, 152, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Cs, P.; Srinath, N.M. Genetic Factors in Nonsyndromic Orofacial Clefts. Glob. Med. Genet. 2020, 7, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Thieme, F.; Ludwig, K.U. The role of noncoding genetic variation in isolated orofacial clefts. J. Dent. Res. 2017, 96, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Iovannisci, D.M.; Yang, W.; Finnell, R.H.; Carmichael, S.L.; Cheng, S.; Lammer, E.J. Endothelial nitric oxide synthase (NOS3) genetic variants, maternal smoking, vitamin use, and risk of human orofacial clefts. Am. J. Epidemiol. 2005, 162, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Beaty, T.H.; Taub, M.A.; Scott, A.F.; Murray, J.C.; Marazita, M.L.; Schwender, H.; Parker, M.; Hetmanski, J.B.; Balakrishnan, P.; Mansilla, M.A.; et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum. Genet. 2013, 132, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Fallin, M.D.; Shi, M.; Ruczinski, I.; Liang, K.Y.; Hetmanski, J.B.; Wang, H.; Ingersoll, R.G.; Huang, S.; Ye, X.; et al. Evidence of gene-environment interaction for the RUNX2 gene and environmental tobacco smoke in controlling the risk of cleft lip with/without cleft palate. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.; Shaffer, J.R.; Deleyiannis, F.; Hecht, J.T.; Wehby, G.L.; Christensen, K.; Feingold, E.; Weinberg, S.M.; Marazita, M.L.; Leslie, E.J. Genome-wide interaction study implicates VGLL2 and alcohol exposure and PRL and smoking in orofacial cleft risk. Front. Cell Dev. Biol. 2022, 10, 621261. [Google Scholar] [CrossRef] [PubMed]
- Haaland, Ø.A.; Lie, R.T.; Romanowska, J.; Gjerdevik, M.; Gjessing, H.K.; Jugessur, A. A genome-wide search for gene-environment effects in isolated cleft lip with or without cleft palate triads points to an interaction between maternal periconceptional vitamin use and variants in ESRRG. Front. Genet. 2018, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Venkataraghavan, S.; Hetmanski, J.B.; Leslie, E.J.; Marazita, M.L.; Feingold, E.; Weinberg, S.M.; Ruczinski, I.; Taub, M.A.; Scott, A.F.; et al. Detecting gene-environment interaction for maternal exposures using case-parent trios ascertained through a case with non-syndromic orofacial cleft. Front. Cell Dev. Biol. 2021, 9, 621018. [Google Scholar] [CrossRef] [PubMed]
- Laurie, C.C.; Doheny, K.F.; Mirel, D.B.; Pugh, E.W.; Bierut, L.J.; Bhangale, T.; Boehm, F.; Caporaso, N.E.; Cornelis, M.C.; Edenberg, H.J.; et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2010, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. minimac2: Faster genotype imputation. Bioinformatics 2015, 31, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J. BinaryDosage: A Package to Create, Merge, and Read Binary Genotype Files. Version 1.0.0. Available online: https://cran.rstudio.com/web/packages/BinaryDosage2020 (accessed on 10 July 2023).
- Morrison, J. GxEScanR: Run GWAS/GWEIS Scans Using Binary Dosage Files [R Package GxEScanR Version 2.0.2]. 2020. Available online: https://cran.r-project.org/web/packages/GxEScanR (accessed on 10 July 2023).
- Gauderman, W.J.; Zhang, P.; Morrison, J.L.; Lewinger, J.P. Finding novel genes by testing G × E interactions in a genome-wide association study. Genet. Epidemiol. 2013, 37, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Gauderman, W.J.; Kim, A.; Conti, D.V.; Morrison, J.; Thomas, D.C.; Vora, H.; Lewinger, J.P. A unified model for the analysis of gene-environment interaction. Am. J. Epidemiol. 2019, 188, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [PubMed]
- Wilderman, A.; van Oudenhove, J.; Kron, J.; Noonan, J.P.; Cotney, J. High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 2018, 23, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Krietenstein, N.; Abraham, S.; Venev, S.V.; Abdennur, N.; Gibcus, J.; Hsieh, T.H.S.; Parsi, K.M.; Yang, L.; Maehr, R.; Mirny, L.A.; et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 2020, 78, 554–565.e7. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Carlson, J.C.; Shaffer, J.R.; Feingold, E.; Wehby, G.; Laurie, C.A.; Jain, D.; Doheny, K.F.; McHenry, T.; Resick, J.; et al. A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum. Mol. Genet. 2016, 25, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Beaty, T.H.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Ruczinski, I.; Hetmanski, J.B.; Liang, K.Y.; Wu, T.; Murray, T.; Fallin, M.D.; et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 2010, 42, 525–529. [Google Scholar] [CrossRef] [PubMed]
- van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- García-Aznar, J.M.; Alvarez, S.A.; del Castillo, T.B. Pivotal role of BCL11B in the immune, hematopoietic and nervous systems: A review of the BCL11B-associated phenotypes from the genetic perspective. Genes. Immun. 2024, 25, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 1, bax028. [Google Scholar] [CrossRef] [PubMed]
- Haaland, Ø.A.; Romanowska, J.; Gjerdevik, M.; Lie, R.T.; Gjessing, H.K.; Jugessur, A. A genome-wide scan of cleft lip triads identifies parent-of-origin interaction effects between ANK3 and maternal smoking, and between ARHGEF10 and alcohol consumption. F1000Research 2019, 8, 960. [Google Scholar] [CrossRef] [PubMed]
- Beaty, T.H.; Ruczinski, I.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Hetmanski, J.B.; Murray, T.; Redett, R.J.; Fallin, M.D.; Liang, K.Y.; et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet. Epidemiol. 2011, 35, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Christensen, K.; Weinberg, C.R.; Romitti, P.; Bathum, L.; Lozada, A.; Morris, R.W.; Lovett, M.; Murray, J.C. Orofacial cleft risk is increased with maternal smoking and specific detoxification-gene variants. Am. J. Hum. Genet. 2007, 80, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Schwender, H.; Ruczinski, I.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Hetmanski, J.B.; Parker, M.M.; Wang, P.; Murray, T.; et al. Evidence of gene−environment interaction for two genes on chromosome 4 and environmental tobacco smoke in controlling the risk of nonsyndromic cleft palate. PLoS ONE 2014, 9, e88088. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakazawa, M.; Kani, S.; Bae, Y.K.; Shimizu, T.; Kageyama, R.; Hibi, M. Zinc finger genes Fezf1 and Fezf2 control neuronal differentiation by repressing Hes5 expression in the forebrain. Development 2010, 137, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Hutchins, B.I.; Ozkan, Y.; Demirel, F.; Stoner, H.; Cheng, P.J.; Esen, I.; Gurbuz, F.; Bicakci, Y.K.; Mengen, E.; et al. Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet. 2014, 95, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Shan, Y.; Whittington, N.C.; Wray, S. Nasal placode development, GnRH neuronal migration and Kallmann Syndrome. Front. Cell Dev. Biol. 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Sun, L.; Song, Y. FEZF1-AS1: A novel vital oncogenic lncRNA in multiple human malignancies. Biosci. Rep. 2019, 39, BSR20191202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Su, F.; Guo, Q.; Tao, X.; Wang, H.; Wang, H.; Li, Q.; Zhang, W. Preeclampsia-associated lncRNA FEZF1-AS1 regulates cell proliferation and apoptosis in placental trophoblast cells through the ELAVL1/NOC2L axis. Cell Div. 2023, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Saunders, G.R.B.; Wang, X.; Chen, F.; Jang, S.K.; Liu, M.; Wang, C.; Gao, S.; Jiang, Y.; Khunsriraksakul, C.; Otto, J.M.; et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature 2022, 612, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Linnér, R.K.; Biroli, P.; Kong, E.; Meddens, S.F.W.; Wedow, R.; Fontana, M.A.; Lebreton, M.; Tino, S.P.; Abdellaoui, A.; Hammerschlag, A.R.; et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 2019, 51, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, M.; Ohn, J.; Seong, R.H.; Chung, J.H.; Kim, K.H.; Jo, S.J.; Kwon, O. Twist2-driven chromatin remodeling governs the postnatal maturation of dermal fibroblasts. Cell Rep. 2022, 39, 110821. [Google Scholar] [CrossRef] [PubMed]
- Marchegiani, S.; Davis, T.; Tessadori, F.; van Haaften, G.; Brancati, F.; Hoischen, A.; Huang, H.; Valkanas, E.; Pusey, B.; Schanze, D.; et al. Recurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes. Am. J. Hum. Genet. 2015, 97, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Merindol, N.; Riquet, A.; Szablewski, V.; Eliaou, J.F.; Puisieux, A.; Bonnefoy, N. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014, 4, e206. [Google Scholar] [CrossRef] [PubMed]
- de Maria, B.; Mazzanti, L.; Roche, N.; Hennekam, R.C. Barber-Say syndrome and Ablepharon-Macrostomia syndrome: An overview. Am. J. Med. Genet. Part A 2016, 170, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Y.; Liu, J.; Wang, Z.; Wu, Q.; Zhang, Z.; Yang, C.J.; Yuan, L.; Ouyang, G. Twist2 contributes to breast cancer progression by promoting an epithelial–mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 2011, 30, 4707–4720. [Google Scholar] [CrossRef] [PubMed]
- Alvizi, L.; Nani, D.; Brito, L.A.; Kobayashi, G.S.; Passos-Bueno, M.R.; Mayor, R. Neural crest E-cadherin loss drives cleft lip/palate by epigenetic modulation via pro-inflammatory gene–environment interaction. Nat. Commun. 2023, 14, 2868. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.L.; Cox, T.C.; Uribe, L.M.M.; Zhu, Y.; Richter, C.T.; Nidey, N.; Standley, J.M.; Deng, M.; Blue, E.; Chong, J.X.; et al. Mutations in the epithelial cadherin-p120-catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 2018, 102, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Green, B.L.; Fasaye, G.A.; Samaranayake, S.G.; Duemler, A.; Gamble, L.A.; Davis, J.L. Frequent cleft lip and palate in families with pathogenic germline CDH1 variants. Front. Genet. 2022, 13, 1012025. [Google Scholar] [CrossRef] [PubMed]
- Bureau, A.; Parker, M.M.; Ruczinski, I.; Taub, M.A.; Marazita, M.L.; Murray, J.C.; Mangold, E.; Noethen, M.M.; Ludwig, K.U.; Hetmanski, J.B.; et al. Whole Exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics 2014, 197, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Di Cello, F.; Flowers, V.L.; Li, H.; Vecchio-Pagán, B.; Gordon, B.; Harbom, K.; Shin, J.; Beaty, R.; Wang, W.; Brayton, C.; et al. Cigarette smoke induces epithelial to mesenchymal transition and increases the metastatic ability of breast cancer cells. Mol. Cancer 2013, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; He, Z.; Yang, X.M.; Li, K.L.; Wang, D.L.; Sun, F.L. RCCD1 depletion attenuates TGF-β-induced EMT and cell migration by stabilizing cytoskeletal microtubules in NSCLC cells. Cancer Lett. 2017, 400, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Sheybani, Z.; Dokoohaki, M.H.; Negahdaripour, M.; Dehdashti, M.; Zolghadr, H.; Moghadami, M.; Masoompour, S.M.; Zolghadr, A.R. The interactions of folate with the enzyme furin: A computational study. RSC Adv. 2021, 11, 23815–23824. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.C.; Lidral, A.C.; McCoy, J.C.; Liu, H.; Cox, L.L.; Zhu, Y.; Anderson, R.D.; Uribe, L.M.M.; Anand, D.; Deng, M.; et al. Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial clefting in humans. Hum. Mutat. 2019, 40, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, T.A.; Phillips, J.B.; Fieg, E.; Bajikar, S.S.; Peirce, J.; Wegner, J.; Luna, A.A.; Fox, E.J.; Yan, Y.L.; Rosenfeld, J.A.; et al. Heterozygous loss-of-function variants significantly expand the phenotypes associated with loss of GDF11. Genet. Med. 2021, 23, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Dicipulo, R.; Norton, K.A.; Fairbridge, N.A.; Kibalnyk, Y.; Fox, S.C.; Hornberger, L.K.; McDermid, H.E. Cecr2 mutant mice as a model for human cat eye syndrome. Sci. Rep. 2021, 11, 3111. [Google Scholar] [CrossRef] [PubMed]
- Fairbridge, N.A.; Dawe, C.E.; Niri, F.H.; Kooistra, M.K.; King-Jones, K.; McDermid, H.E. Cecr2 mutations causing exencephaly trigger misregulation of mesenchymal/ectodermal transcription factors. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, D.; Jin, L.; Zhang, J.; Meng, W.; Jin, L.; Shang, X. The relationship between maternal periconceptional micronutrient supplementation and non-syndromic cleft lip/palate in offspring. Birth Defects Res. 2023, 115, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Jiang, Q.; Liu, L.; Liu, C.; Zhang, Q. Double whammy: The genetic variants in CECR2 and high Hcy on the development of neural tube defects. Front. Genet. 2023, 14, 1189847. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.K.M.; Gordoncillo, N.P.; Atienza, L.M.; Talavera, M.T.M.; Recuenco, M.C. Prevalence and factors associated with folate deficiency among Filipino women of child-bearing age. Malays. J. Nutr. 2020, 26, 229–243. [Google Scholar] [CrossRef]

| Discovery | Replication 1 | Replication 2 | Replication 3 | |||
|---|---|---|---|---|---|---|
| CL/P cases | Controls | CL/P cases | CL/P cases | CL/P cases | ||
| N | 540 | 260 | 137 | 88 | 213 | |
| Maternal vitamin use | Yes | 204 | 222 | 108 | 20 | 36 |
| No | 303 | 38 | 29 | 63 | 170 | |
| Unknown | 33 | 0 | 0 | 5 | 7 | |
| Maternal smoking | Yes | 72 | 23 | 10 | 4 | 33 |
| No | 468 | 237 | 124 | 84 | 180 | |
| Unknown | 0 | 0 | 3 | 0 | 0 | |
| Assigned sex at birth | Males | 320 | 123 | 85 | 59 | 139 |
| Females | 220 | 137 | 52 | 29 | 74 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erdogan-Yildirim, Z.; Carlson, J.C.; Mukhopadhyay, N.; Leslie-Clarkson, E.J.; Padilla, C.D.; Murray, J.C.; Beaty, T.H.; Weinberg, S.M.; Marazita, M.L.; Shaffer, J.R. Gene-by-Environment Interactions Involving Maternal Exposures with Orofacial Cleft Risk in Filipinos. Genes 2025, 16, 876. https://doi.org/10.3390/genes16080876
Erdogan-Yildirim Z, Carlson JC, Mukhopadhyay N, Leslie-Clarkson EJ, Padilla CD, Murray JC, Beaty TH, Weinberg SM, Marazita ML, Shaffer JR. Gene-by-Environment Interactions Involving Maternal Exposures with Orofacial Cleft Risk in Filipinos. Genes. 2025; 16(8):876. https://doi.org/10.3390/genes16080876
Chicago/Turabian StyleErdogan-Yildirim, Zeynep, Jenna C. Carlson, Nandita Mukhopadhyay, Elizabeth J. Leslie-Clarkson, Carmencita D. Padilla, Jeffrey C. Murray, Terri H. Beaty, Seth M. Weinberg, Mary L. Marazita, and John R. Shaffer. 2025. "Gene-by-Environment Interactions Involving Maternal Exposures with Orofacial Cleft Risk in Filipinos" Genes 16, no. 8: 876. https://doi.org/10.3390/genes16080876
APA StyleErdogan-Yildirim, Z., Carlson, J. C., Mukhopadhyay, N., Leslie-Clarkson, E. J., Padilla, C. D., Murray, J. C., Beaty, T. H., Weinberg, S. M., Marazita, M. L., & Shaffer, J. R. (2025). Gene-by-Environment Interactions Involving Maternal Exposures with Orofacial Cleft Risk in Filipinos. Genes, 16(8), 876. https://doi.org/10.3390/genes16080876