
The Role of MLPA in Detecting Syndromic Submicroscopic Copy Number Variations in Normal QF-PCR Miscarriage Specimens

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Inclusion
2.2. Multiplex Ligation-Dependent Probe Amplification (MLPA) Analysis
3. Results
3.1. Abnormalities Detected in Early Pregnancy Loss (EPL) Group
3.2. Abnormalities Detected in Late Pregnancy Loss (LPL) Group
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MLPA | Multiplex ligation-dependent probe amplification |
| CNV | Copy number variation |
| DNA | Deoxyribonucleic acid |
| QF-PCR | Quantitative fluorescent-polymerase chain reaction |
| ASRM | The American Society for Reproductive Medicine |
| ACOG | The American College of Obstetricians and Gynecologists |
| EPL | Early pregnancy loss |
| LPL | Late pregnancy loss |
| pCNV | Pathogenic copy number variation |
| lpCNV | Likely pathogenic copy number variation |
| POC | Product of conception |
| NF1 | Neurofibromatosis type 1 |
| PCR | Polymerase chain reaction |
| LETM1 | Leucine zipper and EF-hand containing transmembrane protein 1 |
| WHS | Wolf–Hirschhorn syndrome |
| WHSC1 | Wolf–Hirschhorn syndrome candidate 1 |
| WHSCR | Wolf–Hirschhorn syndrome critical region |
| GATA3 | GATA binding protein 3 |
| RSTS1 | Rubinstein–Taybi syndrome type 1 |
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 |
| MBD5 | Methyl-CpG binding domain protein 5 |
| SATB2 | SATB homeobox 2 |
| CREBBP | CREB binding lysine acetyltransferase |
| RABL2B | RAB, member of RAS oncogene family like 2B |
| MECP2 | Methyl-CpG binding protein 2 |
| HDRS | Hypoparathyroidism, sensorineural deafness, and renal insufficiency syndrome |
| DGS | DiGeorge syndrome |
| VCFS | Velocardiofacial syndrome |
| RTS | Rett syndrome |
| PHMDS | Phelan–McDermid syndrome |
| CMA | Chromosomal microarray |
| CNV-seq | Copy number variation sequencing |
| aCGH | Array comparative genomic hybridization |
| SNP | Single-nucleotide polymorphism |
| UPD | Uniparental disomy |
| NGS | Next-generation sequencing |
References
- Quenby, S.; Gallos, I.D.; Dhillon-Smith, R.K.; Podesek, M.; Stephenson, M.D.; Fisher, J.; Brosens, J.J.; Brewin, J.; Ramhorst, R.; Lucas, E.S.; et al. Miscarriage matters: The epidemiological, physical, psychological, and economic costs of early pregnancy loss. Lancet 2021, 397, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Practice Committee of the American Society for Reproductive Medicine. Evaluation and treatment of recurrent pregnancy loss: A committee opinion. Fertil. Steril. 2012, 98, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin No. 200: Early Pregnancy Loss. Obstet. Gynecol. 2018, 132, e197–e207. [Google Scholar] [CrossRef] [PubMed]
- Mehra, V.M.; Farooqi, S.; Sriram, P.; Tunde-Byass, M. Diagnosis and management of early pregnancy loss. CMAJ 2024, 196, E1162–E1168. [Google Scholar] [CrossRef] [PubMed]
- Odendaal, H.J. Strong Association Between Placental Pathology and Second-trimester Miscarriage. Arch. Obstet. Gynaecol. 2021, 2, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Genovese, H.G.; McQueen, D.B. The prevalence of sporadic and recurrent pregnancy loss. Fertil. Steril. 2023, 120, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, A.J.; Weinberg, C.R.; O’Connor, J.F.; Baird, D.D.; Schlatterer, J.P.; Canfield, R.E.; Armstrong, E.G.; Nisula, B.C. Incidence of early loss of pregnancy. N. Engl. J. Med. 1988, 319, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, C.; Wang, L.; Chen, D.; Guang, W.; French, J. Conception, early pregnancy loss, and time to clinical pregnancy: A population-based prospective study. Fertil. Steril. 2003, 79, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Zinaman, M.J.; Clegg, E.D.; Brown, C.C.; O’Connor, J.; Selevan, S.G. Estimates of human fertility and pregnancy loss. Fertil. Steril. 1996, 65, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Annual Capri Workshop Group. Early pregnancy loss: The default outcome for fertilized human oocytes. J. Assist. Reprod. Genet. 2020, 37, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Farren, J.; Mitchell-Jones, N.; Verbakel, J.Y.; Timmerman, D.; Jalmbrant, M.; Bourne, T. The psychological impact of early pregnancy loss. Hum. Reprod. Update 2018, 24, 731–749. [Google Scholar] [CrossRef] [PubMed]
- Due, C.; Chiarolli, S.; Riggs, D.W. The impact of pregnancy loss on men’s health and wellbeing: A systematic review. BMC Pregnancy Childbirth 2017, 17, 380. [Google Scholar] [CrossRef] [PubMed]
- Mróz, M.; Bień, A.; Iwanowicz-Palus, G.; Krysa, J. Identification of Factors Affecting Self-Efficacy in Women with Spontaneous Pregnancy Loss. Healthcare 2023, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Strumpf, E.C.; Austin, N.; Lang, A.; Derksen, S.; Bolton, J.; Brownell, M.; Gregory, P.; Chateau, D.; Heaman, M. The effects of early pregnancy loss on health outcomes and health care utilization and costs. Health Serv. Res. 2022, 57, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Coomarasamy, A.; Gallos, I.D.; Papadopoulou, A.; Dhillon-Smith, R.K.; Al-Memar, M.; Brewin, J.; Christiansen, O.B.; Stephenson, M.D.; Oladapo, O.T.; Wijeyaratne, C.N.; et al. Sporadic miscarriage: Evidence to provide effective care. Lancet 2021, 397, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Essers, R.; Lebedev, I.N.; Kurg, A.; Fonova, E.A.; Stevens, S.J.C.; Koeck, R.M.; von Rango, U.; Brandts, L.; Deligiannis, S.P.; Nikitina, T.V.; et al. Prevalence of chromosomal alterations in first-trimester spontaneous pregnancy loss. Nat. Med. 2023, 29, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.; Kalleas, D.; Cooney, D.; Lamb, M.; Lister, L. Meiosis and maternal aging: Insights from aneuploid oocytes and trisomy births. Cold Spring Harb. Perspect. Biol. 2015, 7, a017970. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Allanson, B.; Jennings, B.; Jacques, A.; Charles, A.K.; Keil, A.D.; Dickinson, J.E. Infection and fetal loss in the mid-second trimester of pregnancy. Aust. N. Z. J. Obstet. Gynaecol. 2010, 50, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; Taioli, E.; Cetin, I.; Marinoni, A.; Gerosa, S.; Villa, M.V.; Bozzo, M.; Mannucci, P.M. Mutations in coagulation factors in women with unexplained late fetal loss. N. Engl. J. Med. 2000, 343, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.; Sobczyk, K.; Elebert, R.; Tarleton, D.; Casey, B.; Doyle, S.; Crosby, D.; Allen, C. Second-trimester miscarriage: A review of postnatal investigations and subsequent pregnancy outcomes. Ir. J. Med. Sci. 2023, 192, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Michels, T.C.; Tiu, A.Y. Second trimester pregnancy loss. Am. Fam. Physician 2007, 76, 1341–1346. [Google Scholar] [PubMed]
- Simpson, J.L. Causes of fetal wastage. Clin. Obstet. Gynecol. 2007, 50, 10–30. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Lauer, S.; Gresham, D. An evolving view of copy number variants. Curr. Genet. 2019, 65, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Li, X.; Li, J.; Chen, H.; Liu, L.; Zhou, S.; Xu, Z. Sequential application of copy number variation sequencing and quantitative fluorescence polymerase chain reaction in genetic analysis of miscarriage and stillbirth. Mol. Genet. Genom. Med. 2023, 11, e2187. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, Q.; Zhang, X.; Yu, Z.; Zhong, Z. Analysis of Genomic Copy Number Variation in Miscarriages During Early and Middle Pregnancy. Front. Genet. 2021, 12, 732419. [Google Scholar] [CrossRef] [PubMed]
- Schouten, J.; van Vught, P.; Galjaard, R.J. Multiplex Ligation-Dependent Probe Amplification (MLPA) for Prenatal Diagnosis of Common Aneuploidies. Methods Mol. Biol. 2019, 1885, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int. J. Mol. Sci. 2012, 13, 3245–3276. [Google Scholar] [CrossRef] [PubMed]
- Eijk-Van Os, P.G.; Schouten, J.P. Multiplex Ligation-dependent Probe Amplification (MLPA®) for the detection of copy number variation in genomic sequences. Methods Mol. Biol. 2011, 688, 97–126. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.S.; van den Veyver, I.; Eng, C.M. Multiplex ligation-dependent probe amplification (MLPA) and prenatal diagnosis. Prenat. Diagn. 2012, 32, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, A.S.; Darbro, B.W. A comprehensive list of human microdeletion and microduplication syndromes. BMC Genom. Data 2022, 23, 82. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Lecce, R.; Fischetto, R.; Murdolo, M.; Faravelli, F.; Selicorni, A.; Buttè, C.; Memo, L.; Capovilla, G.; Neri, G. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am. J. Hum. Genet. 2003, 72, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.J.; Ricke, D.O.; Denison, K.; Abmayr, S.; Cotter, P.D.; Hirschhorn, K.; Keinänen, M.; McDonald-McGinn, D.; Somer, M.; Spinner, N.; et al. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum. Mol. Genet. 1997, 6, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Doronzio, P.N. Dissecting the Wolf-Hirschhorn syndrome phenotype: WHSC1 is a neurodevelopmental gene contributing to growth delay, intellectual disability, and to the facial dysmorphism. J. Hum. Genet. 2018, 63, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Hannes, F.; Drozniewska, M.; Vermeesch, J.R.; Haus, O. Duplication of the Wolf-Hirschhorn syndrome critical region causes neurodevelopmental delay. Eur. J. Med. Genet. 2010, 53, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Roselló, M.; Monfort, S.; Orellana, C.; Ferrer-Bolufer, I.; Quiroga, R.; Oltra, S.; Martínez, F. Submicroscopic duplication of the Wolf-Hirschhorn critical region with a 4p terminal deletion. Cytogenet. Genome Res. 2009, 125, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Cheung, S.W.; Breman, A.M.; Bacino, C.A. 4p16.3 microdeletions and microduplications detected by chromosomal microarray analysis: New insights into mechanisms and critical regions. Am. J. Med. Genet. A 2016, 170, 2540–2550. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Béna, F.; Breckpot, J.; Philip, N.; Menten, B.; Van Esch, H.; Scalais, E.; Salamone, J.M.; Fong, C.T.; Kussmann, J.L.; et al. Duplications of the critical Rubinstein-Taybi deletion region on chromosome 16p13.3 cause a novel recognisable syndrome. J. Med. Genet. 2010, 47, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Demeer, B.; Andrieux, J.; Receveur, A.; Morin, G.; Petit, F.; Julia, S.; Plessis, G.; Martin-Coignard, D.; Delobel, B.; Firth, H.V.; et al. Duplication 16p13.3 and the CREBBP gene: Confirmation of the phenotype. Eur. J. Med. Genet. 2013, 56, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Socha, M.; Szoszkiewicz, A.; Simon, D.; Jamsheer, A. A pure de novo 16p13.3 duplication and amplification in a patient with femoral hypoplasia, psychomotor retardation, heart defect, and facial dysmorphism-a case report and literature review of the partial 16p13.3 trisomy syndrome. J. Appl. Genet. 2023, 64, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Chiara, P.; Melissa, A.; Elisena, M.; Paolo, G.-F.; Angelika, M.; Francesco, C.; Giandomenico, P.; Giuseppe, C. 16p13.3 microduplication syndrome: A new characteristic case without intellectual disability. Gene Rep. 2016, 4, 218–221. [Google Scholar] [CrossRef]
- Ponnala, R.; Ranganath, P.; Dutta, U.R.; Pidugu, V.K.; Dalal, A.B. Phenotypic and molecular characterization of partial trisomy 2q resulting from insertion-duplication in chromosome 18q: A case report and review of literature. Cytogenet. Genome Res. 2012, 136, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Osaka, H.; Miyake, N.; Fujita, A.; Uchiyama, Y.; Seyama, R.; Koshimizu, E.; Miyatake, S.; Mizuguchi, T.; Takeda, S.; et al. Distal 2q duplication in a patient with intellectual disability. Hum. Genome Var. 2022, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Plessis, G.; Couturier, J.; Turleau, C.; Despoisses, S.; Delavenne, J. ‘Pure’ partial trisomy 2q in a male owing to malsegregation of a maternal translocation t(X;2)(p22.3;q32.1). J. Med. Genet. 1985, 22, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Kiholm Lund, A.B.; Hove, H.D.; Kirchhoff, M. A 15q24 microduplication, reciprocal to the recently described 15q24 microdeletion, in a boy sharing clinical features with 15q24 microdeletion syndrome patients. Eur. J. Med. Genet. 2008, 51, 520–526. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Smolarek, T.A.; Walker, M.E.; Schorry, E.K.; Immken, L.L.; Patel, G.; Abbott, M.A.; Lanpher, B.C.; Ou, Z.; Kang, S.H.; et al. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum. Genet. 2009, 126, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, S.; Lichtner, P.; Peykar-Derakhshandeh, P.; Murken, J.; Haas, O.A.; Back, E.; Wolff, G.; Zabel, B.; Barisic, I.; Rauch, A.; et al. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGS2). Eur. J. Hum. Genet. 1998, 6, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Groenen, P.; Nesbit, M.A.; Schuffenhauer, S.; Lichtner, P.; Vanderlinden, G.; Harding, B.; Beetz, R.; Bilous, R.W.; Holdaway, I.; et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 2000, 406, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Muroya, K.; Hasegawa, T.; Ito, Y.; Nagai, T.; Isotani, H.; Iwata, Y.; Yamamoto, K.; Fujimoto, S.; Seishu, S.; Fukushima, Y.; et al. GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. J. Med. Genet. 2001, 38, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Belge, H.; Dahan, K.; Cambier, J.F.; Benoit, V.; Morelle, J.; Bloch, J.; Vanhille, P.; Pirson, Y.; Demoulin, N. Clinical and mutational spectrum of hypoparathyroidism, deafness and renal dysplasia syndrome. Nephrol. Dial. Transplant. 2017, 32, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Adachi, M.; Takahashi, H.; Shirakura, M.; Honkura, Y.; Yamauchi, D.; Katori, Y. HDR syndrome, detected in the neonatal period by newborn hearing screening. Auris Nasus Larynx 2024, 51, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Fukami, M.; Muroya, K.; Miyake, T.; Iso, M.; Kato, F.; Yokoi, H.; Suzuki, Y.; Tsubouchi, K.; Nakagomi, Y.; Kikuchi, N.; et al. GATA3 abnormalities in six patients with HDR syndrome. Endocr. J. 2011, 58, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lichtner, P.; König, R.; Hasegawa, T.; Van Esch, H.; Meitinger, T.; Schuffenhauer, S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J. Med. Genet. 2000, 37, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, L.; Sinibaldi, L.; Capalbo, A.; Bottillo, I.; Mancuso, B.; Torres, B.; Novelli, A.; Digilio, M.C.; Dallapiccola, B. HDR (Hypoparathyroidism, Deafness, Renal dysplasia) syndrome associated to GATA3 gene duplication. Clin. Genet. 2009, 76, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Clinical Genome Resource. Available online: https://search.clinicalgenome.org/kb/gene-dosage/GATA3 (accessed on 20 June 2025).
- Vidal, S.; Pascual-Alonso, A.; Rabaza-Gairí, M.; Gerotina, E.; Brandi, N.; Pacheco, P.; Xiol, C.; Pineda, M.; Armstrong, J. Characterization of large deletions of the MECP2 gene in Rett syndrome patients by gene dosage analysis. Mol. Genet. Genomic Med. 2019, 7, e793. [Google Scholar] [CrossRef] [PubMed]
- Gold, W.A.; Percy, A.K.; Neul, J.L.; Cobb, S.R.; Pozzo-Miller, L.; Issar, J.K.; Ben-Zeev, B.; Vignoli, A.; Kaufmann, W.E. Rett syndrome. Nat. Rev. Dis. Primers 2024, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Di Filippo, T.; Roccella, M. The Quality of Life in Girls with Rett Syndrome. Ment. Illn. 2016, 8, 6302. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Christodoulou, J. MECP2 Disorders. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1497/ (accessed on 11 February 2025).
- Zwanenburg, R.J.; Ruiter, S.A.; van den Heuvel, E.R.; Flapper, B.C.; Van Ravenswaaij-Arts, C.M. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: A systematic and prospective study in 34 children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Levy, T.; Foss-Feig, J.H.; Betancur, C.; Siper, P.M.; Trelles-Thorne, M.D.P.; Halpern, D.; Frank, Y.; Lozano, R.; Layton, C.; Britvan, B.; et al. Strong evidence for genotype-phenotype correlations in Phelan-McDermid syndrome: Results from the developmental synaptopathies consortium. Hum. Mol. Genet. 2022, 31, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Sun, M.; Zhao, Y.; Fu, Y.; Yuan, H.; Dai, Y.; Liang, F.; He, Y.; Liu, Y. Analysis of tissue from pregnancy loss and aborted fetus with ultrasound anomaly using subtelomeric MLPA and chromosomal array analysis. J. Matern. Fetal Neonatal Med. 2022, 35, 3064–3069. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, D.; O’Donoghue, K. 572: The use of multiplex ligation-dependent probe amplification (MLPA) versus karyotype analysis in the investigation of pregnancy loss. Am. J. Obstet. Gynecol. 2013, 208, S245. [Google Scholar] [CrossRef]
- Caramins, M.C.; Saville, T.; Shakeshaft, R.; Mullan, G.L.; Miller, B.; Yip, M.Y.; Buckley, M.F. A comparison of molecular and cytogenetic techniques for the diagnosis of pregnancy loss. Genet. Med. 2011, 13, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Bozhinovski, G.; Terzikj, M.; Kubelka-Sabit, K.; Jasar, D.; Lazarevski, S.; Livrinova, V.; Plaseska-Karanfilska, D. Chromosomal Abnormalities in Early Pregnancy Losses: A Study of 900 Samples. Balkan J. Med. Genet. 2023, 26, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Isidori, I.; Spapperi, C.; Barbati, A.; Mencarelli, A.; Stangoni, G. QF-PCR and MLPA: A reliable molecular system to detect chromosomal alterations in miscarriages. Clin. Exp. Obstet. Gynecol. 2017, 44, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Zimowski, J.G.; Massalska, D.; Pawelec, M.; Bijok, J.; Michałowska, A.; Roszkowski, T. First-trimester spontaneous pregnancy loss-molecular analysis using multiplex ligation-dependent probe amplification. Clin. Genet. 2016, 89, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lyu, S.W.; Sung, S.R.; Park, J.E.; Cha, D.H.; Yoon, T.K.; Ko, J.J.; Shim, S.H. Molecular analysis of miscarriage products using multiplex ligation-dependent probe amplification (MLPA): Alternative to conventional karyotype analysis. Arch. Gynecol. Obstet. 2015, 291, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Qi, H.; Du, Y.; Cai, L.; Wen, X.; Wan, Q.; Luo, Y.; Zhu, J. Analysis of potential copy-number variations and genes associated with first-trimester missed abortion. Heliyon 2023, 9, e18868. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Zhou, Q.; Chai, H.; Wen, J.; Zhao, H.; Taylor, H.S.; Jiang, Y.H.; Li, P. Estimation on risk of spontaneous abortions by genomic disorders from a meta-analysis of microarray results on large case series of pregnancy losses. Mol. Genet. Genomic Med. 2023, 11, e2181. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Tucker, M.E.; Escobar, L.F.; Neill, N.J.; Torchia, B.S.; McDaniel, L.D.; Schultz, R.A.; Chong, K.; Chitayat, D. Diagnostic utility of microarray testing in pregnancy loss. Ultrasound Obstet. Gynecol. 2015, 46, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Maisenbacher, M.K.; Merrion, K.; Levy, B.; Kutteh, W.H. Single nucleotide polymorphism (SNP) array analysis of 63,277 products of conception (POC) samples: A 10-year laboratory experience. Fertil. Steril. 2020, 114, e47. [Google Scholar] [CrossRef]
- Xue, H.; Guo, Q.; Yu, A.; Lin, M.; Chen, X.; Xu, L. Genetic analysis of chorionic villus tissues in early missed abortions. Sci. Rep. 2023, 13, 21719. [Google Scholar] [CrossRef] [PubMed]
- Drozdov, G.V.; Kashevarova, A.A.; Lebedev, I.N. Copy number variations in spontaneous abortions: A meta-analysis. J. Assist. Reprod. Genet. 2025, 42, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Dahdouh, E.M.; Kutteh, W.H. Genetic testing of products of conception in recurrent pregnancy loss evaluation. Reprod. Biomed. Online 2021, 43, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Keren, B. The advantages of SNP arrays over CGH arrays. Mol. Cytogenet. 2014, 7, I31. [Google Scholar] [CrossRef] [PubMed]
- Dugoff, L.; Norton, M.E.; Kuller, J.A. The use of chromosomal microarray for prenatal diagnosis. Am. J. Obstet. Gynecol. 2016, 215, B2–B9. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Dzidic, N.; Strecker, M.N.; Commander, S.; Travis, M.K.; Doherty, C.; Tyson, R.W.; Mendoza, A.E.; Stephenson, M.; Dise, C.A.; et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: Outcomes, benefits, and challenges. Genet. Med. 2017, 19, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.A.J.; van Maarle, M.; Hamer, G.; Mastenbroek, S.; Goddijn, M.; van Wely, M. Cytogenetic testing of pregnancy loss tissue: A meta-analysis. Reprod. Biomed. Online 2020, 40, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Regan, L.; Rai, R.; Saravelos, S.; Li, T.C. Recurrent MiscarriageGreen-top Guideline No. 17. BJOG Int. J. Obstet. Gynaecol. 2023, 130, e9–e39. [Google Scholar] [CrossRef] [PubMed]
- Bender Atik, R.; Christiansen, O.B.; Elson, J.; Kolte, A.M.; Lewis, S.; Middeldorp, S.; McHeik, S.; Peramo, B.; Quenby, S.; Nielsen, H.S.; et al. ESHRE guideline: Recurrent pregnancy loss: An update in 2022. Hum. Reprod. Open 2023, 2023, hoad002. [Google Scholar] [CrossRef] [PubMed]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Su, L.; Xie, X.; He, D.; Chen, X.; Wang, M.; Wang, L.; Zheng, L.; Xu, L. Comprehensive analysis of early pregnancy loss based on cytogenetic findings from a tertiary referral center. Mol. Cytogenet. 2021, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, R.; Jiang, L.; Meng, L.; Tan, J.; Qiao, F.; Wang, Y.; Zhang, C.; Cheng, Q.; Jiang, Z.; et al. Identification of Chromosomal Abnormalities in Early Pregnancy Loss Using a High-Throughput Ligation-Dependent Probe Amplification-Based Assay. J. Mol. Diagn. 2021, 23, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Donaghue, C.; Davies, N.; Ahn, J.W.; Thomas, H.; Ogilvie, C.M.; Mann, K. Efficient and cost-effective genetic analysis of products of conception and fetal tissues using a QF-PCR/array CGH strategy; five years of data. Mol. Cytogenet. 2017, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Christofolini, D.M.; Bevilacqua, L.B.; Mafra, F.A.; Kulikowski, L.D.; Bianco, B.; Barbosa, C.P. Genetic analysis of products of conception. Should we abandon classic karyotyping methodology? Einstein 2021, 19, eAO5945. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Peng, Y.; Lv, W.; Deng, L.; Zhang, Y.; Li, H.; Yang, P.; Zhang, J.; Song, Z.; Xu, G.; et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J. Mol. Diagn. 2014, 16, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Yang, S.; Cheng, L.; Duan, J.; Li, J.; Kang, J.; Wang, F.; Liu, J.; Zheng, F.; Ma, J.; et al. Identification of chromosomal abnormalities in miscarriages by CNV-Seq. Mol. Cytogenet. 2024, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Zhang, Q.; Lin, Z.; Ye, J.; Shen, X.; Zhou, L.; Cai, W. Analysis of copy number variations and possible candidate genes in spontaneous abortion by copy number variation sequencing. Front. Endocrinol. 2023, 14, 1218793. [Google Scholar] [CrossRef] [PubMed]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-Generation Sequencing Technologies. Cold Spring Harb. Perspect. Med. 2019, 9, a036798. [Google Scholar] [CrossRef] [PubMed]
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, J.; Wu, Y.; Shi, X.; Xin, X.; Li, S.; Zeng, W.; Deng, D.; Feng, L.; Chen, S.; et al. Analysis of Chromosomal Copy Number in First-Trimester Pregnancy Loss Using Next-Generation Sequencing. Front. Genet. 2020, 11, 545856. [Google Scholar] [CrossRef] [PubMed]
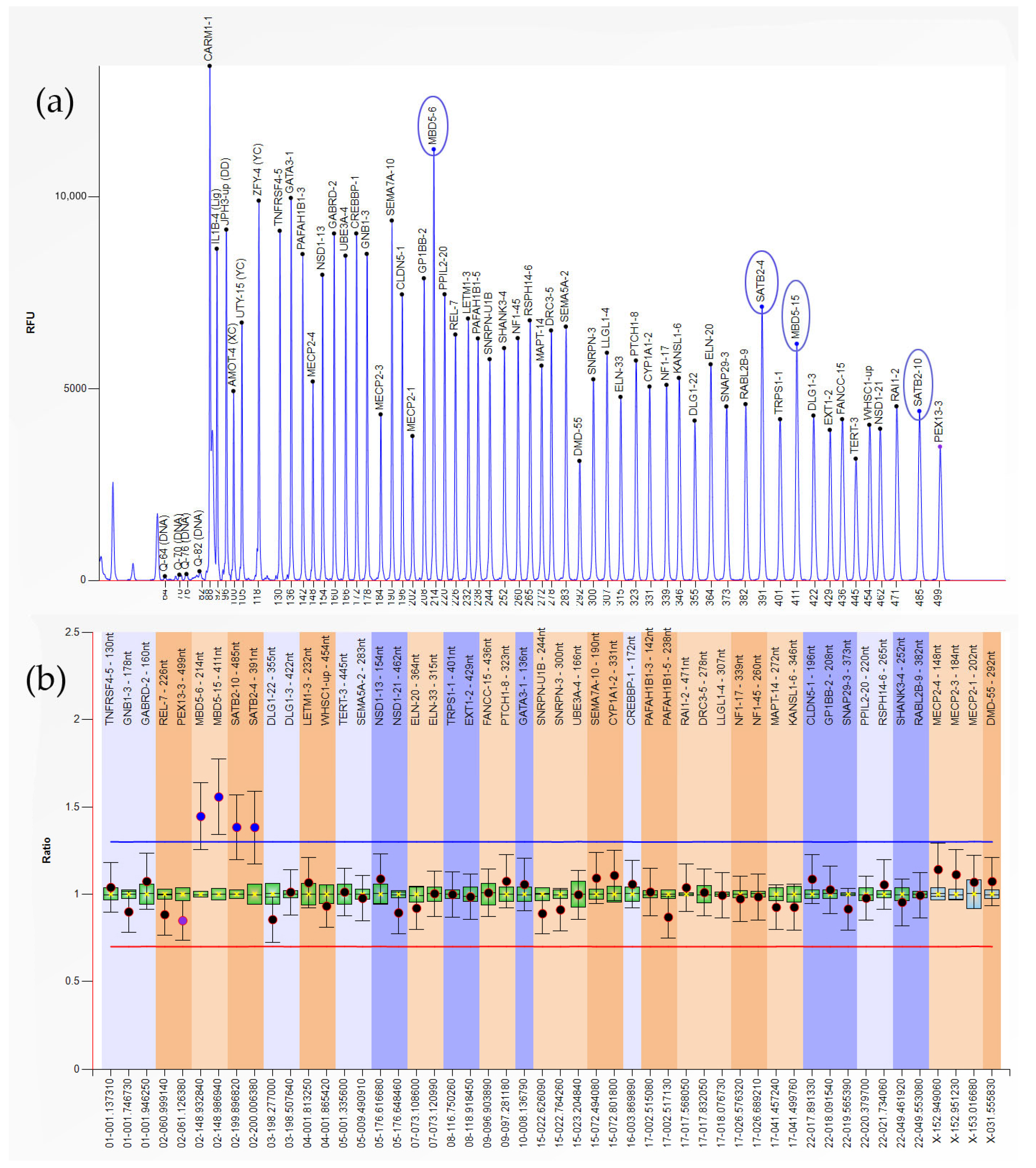
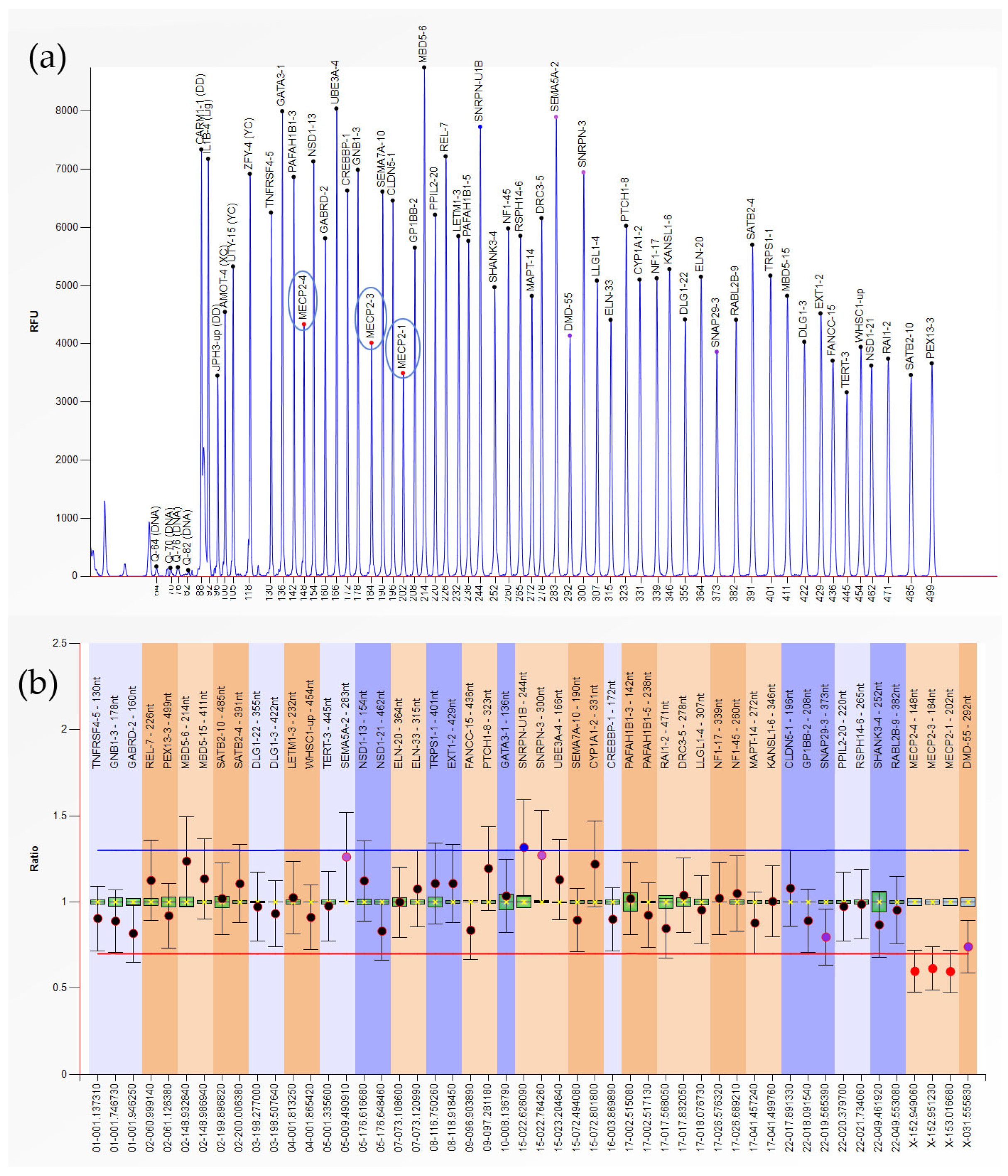

{kind=link}
{kind=link}
| Results | ||
|---|---|---|
| Positive n/% * | Negative n/% * | |
| Early Pregnancy Loss (EPL) | 10 (11.9%) | 51 (60.7%) |
| Late Pregnancy Loss (LPL) | 1 (1.2%) | 22 (26.2%) |
| Total n/% * | 11 (13.1%) | 73 (86.9%) |
| Type of Abnormality | n | Gene-Exon (Locus) |
|---|---|---|
| Heterozygous Duplication | 1 | MBD5-6,15; SATB2-4,10 (2q23.1-2q33.1) |
| 3 | LETM1-3; WHSC1-up (4p16.3) | |
| 1 | GATA3-1 (10p14) | |
| 1 | CYP1A1-2 (15q24.1) | |
| 1 | CREBBP-1 (16p13.3) | |
| Heterozygous Deletion | 1 | GATA3-1 (10p14) |
| 1 | RABL2B-9 (22q13.33) | |
| Hemizygous Deletion | 1 | MECP2-1,3,4 (Xq28) |
| Study | MLPA Assay | Analyzed Cases (n) | Structural Abnormality/CNV (%) |
|---|---|---|---|
| Present study | microdeletion syndromes | 84 | 13.1% (pCNV–3.6%) |
| Caramins et al., 2011 [70] | subtelomeric | 284 | 2.5% |
| O’Leary et al., 2013 [69] | unspecified | 102 | 7.8% * |
| Kim et al., 2014 [74] | subtelomeric | 347 | 3.5% |
| Zimowski et al., 2016 [73] | subtelomeric/subcentromeric | 181 | 7.2% |
| Isidori et al., 2017 [72] | subtelomeric | 264 | 1.5% |
| Lou et al., 2020 [68] | subtelomeric | 172 | 4.1% |
| Bozhinovski et al., 2024 [71] | subtelomeric | 768 | 3.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu-Hobeanu, G.; Cucu, M.-G.; Calotă-Dobrescu, A.; Dragotă, L.; Riza, A.-L.; Streață, I.; Pleșea, R.M.; Pătru, C.L.; Comănescu, C.M.; Tudorache, Ș.; et al. The Role of MLPA in Detecting Syndromic Submicroscopic Copy Number Variations in Normal QF-PCR Miscarriage Specimens. Genes 2025, 16, 867. https://doi.org/10.3390/genes16080867
Popescu-Hobeanu G, Cucu M-G, Calotă-Dobrescu A, Dragotă L, Riza A-L, Streață I, Pleșea RM, Pătru CL, Comănescu CM, Tudorache Ș, et al. The Role of MLPA in Detecting Syndromic Submicroscopic Copy Number Variations in Normal QF-PCR Miscarriage Specimens. Genes. 2025; 16(8):867. https://doi.org/10.3390/genes16080867
Chicago/Turabian StylePopescu-Hobeanu, Gabriela, Mihai-Gabriel Cucu, Alexandru Calotă-Dobrescu, Luminița Dragotă, Anca-Lelia Riza, Ioana Streață, Răzvan Mihail Pleșea, Ciprian Laurențiu Pătru, Cristina Maria Comănescu, Ștefania Tudorache, and et al. 2025. "The Role of MLPA in Detecting Syndromic Submicroscopic Copy Number Variations in Normal QF-PCR Miscarriage Specimens" Genes 16, no. 8: 867. https://doi.org/10.3390/genes16080867
APA StylePopescu-Hobeanu, G., Cucu, M.-G., Calotă-Dobrescu, A., Dragotă, L., Riza, A.-L., Streață, I., Pleșea, R. M., Pătru, C. L., Comănescu, C. M., Tudorache, Ș., Iliescu, D., & Burada, F. (2025). The Role of MLPA in Detecting Syndromic Submicroscopic Copy Number Variations in Normal QF-PCR Miscarriage Specimens. Genes, 16(8), 867. https://doi.org/10.3390/genes16080867