
Construction of Gene Regulatory Networks Based on Spatial Multi-Omics Data and Application in Tumor-Boundary Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Single-Cell and Spatial-Transcriptomics Data Collection
2.2. Dimension Reduction and Clustering Analysis of Single-Cell Transcriptome Data
2.3. Identification of Malignant Cells from scRNA-Seq Data
2.4. Spatial-Transcriptomics Data Analysis
2.5. Spatial Cell Communication Strength Analysis
2.6. Definition of Tumor-Boundary-Based Spatial-Transcriptome Data
2.7. Flow for Constructing Spatial Gene Regulatory Networks
2.8. Spatial-Proteomic Profiling Analysis
2.9. Enrichment Analysis and Survival Analysis
3. Results
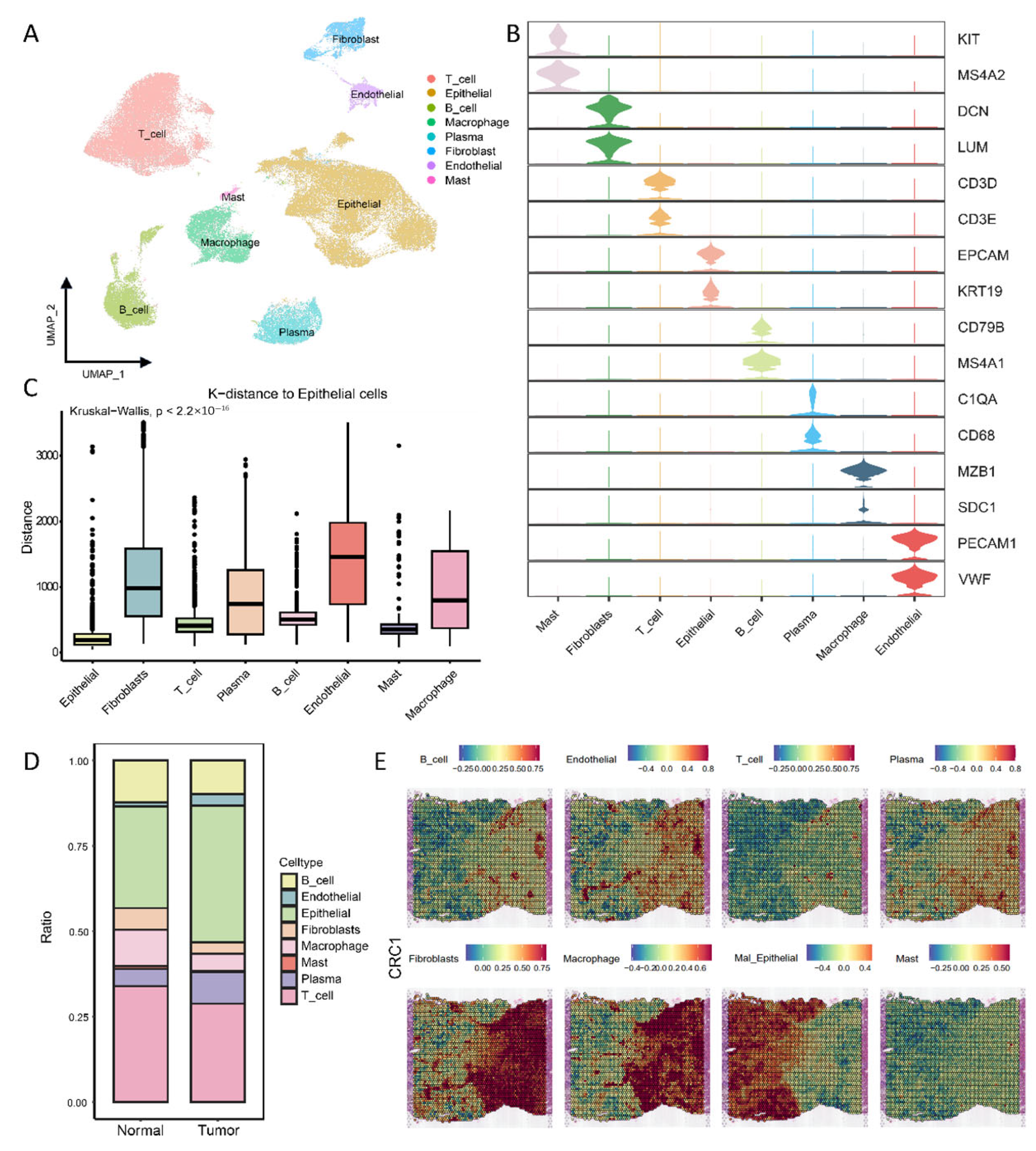
3.1. Single-Cell and Spatial-Transcriptome Profiles of Colorectal Cancer
3.2. Tumor-Boundary Cell Interactions
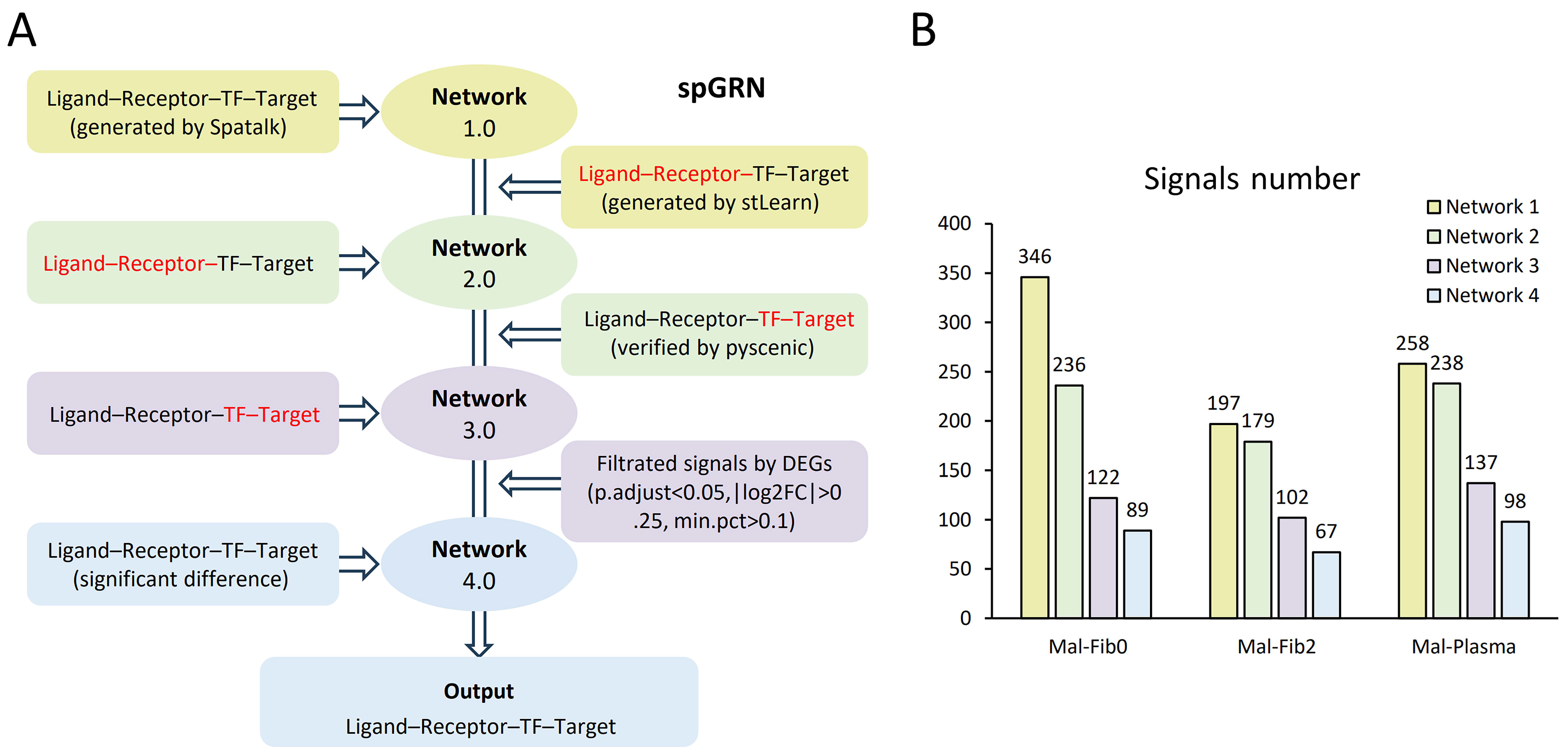
3.3. Spatial-Resolved Gene-Regulatory Network Construction at the Cell Tumor Boundary
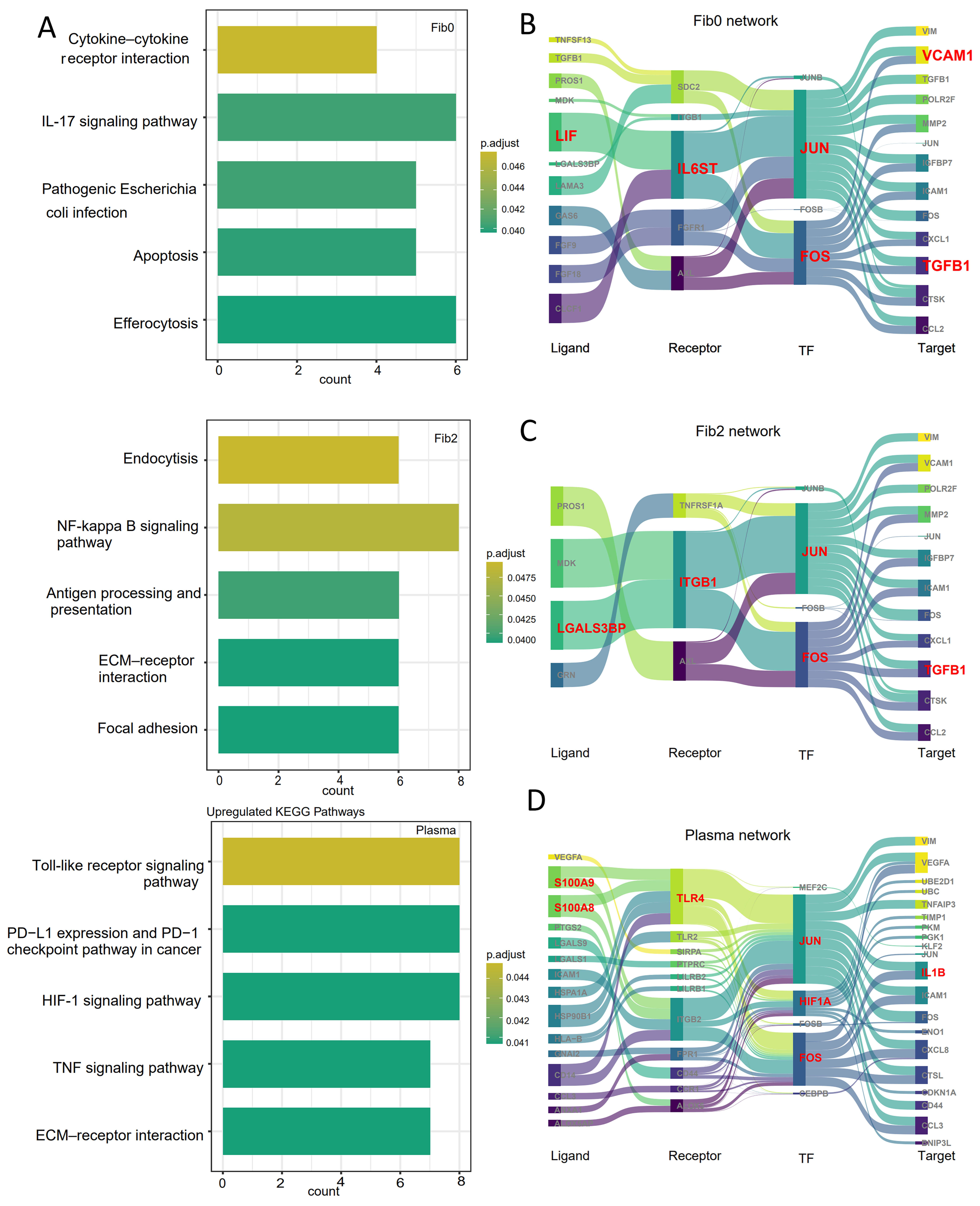
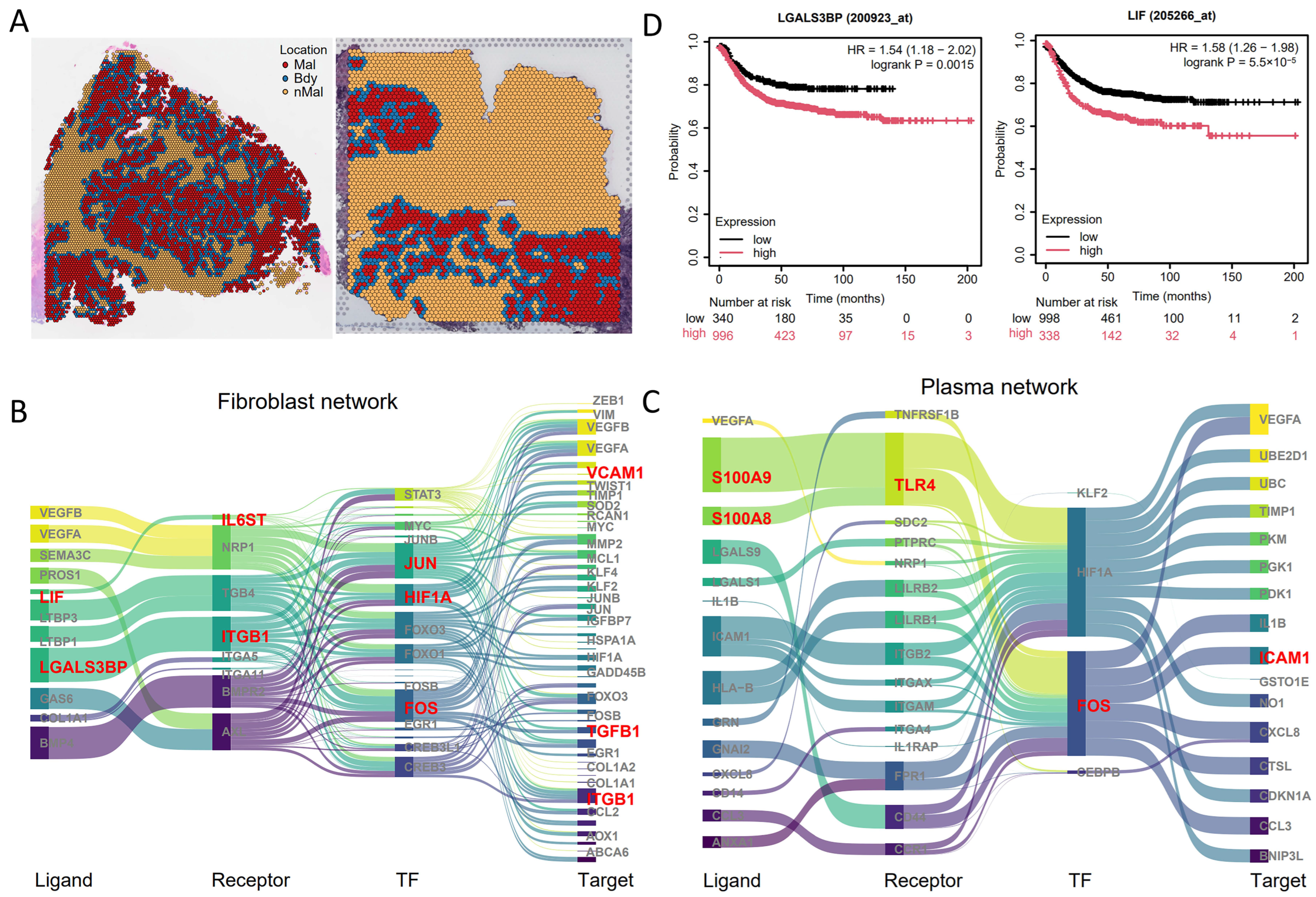
3.4. spGRN Analysis of Plasma/Fibroblast Cells and Malignant Cells at the Spatial Boundary
3.5. Independent Validation of Main Signaling Molecules in CRC
3.6. Validation of the spGRN Results Using Spatial-Proteomics Data
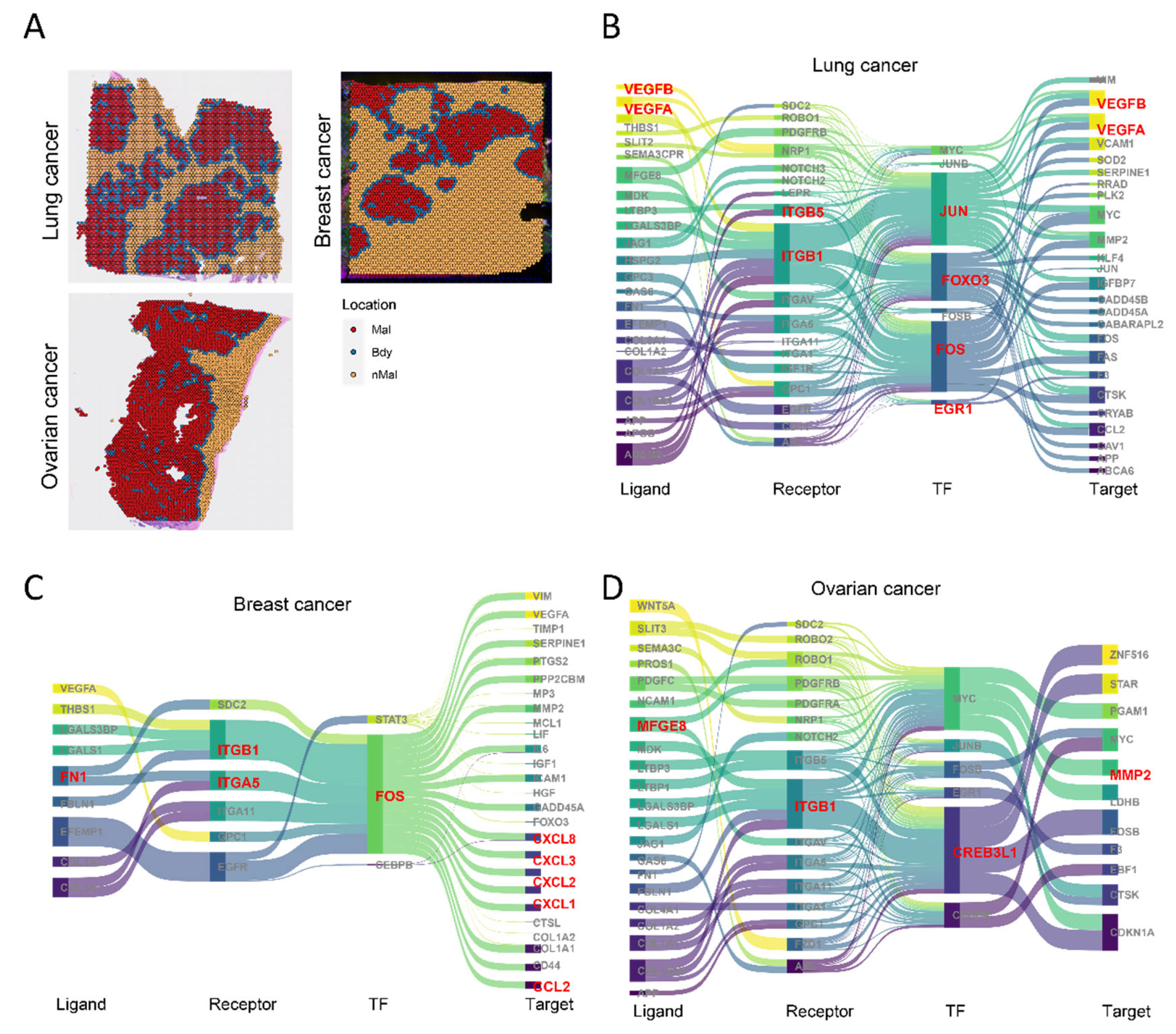
3.7. Reproducible Regulatory Networks Identified in Pan-Cancer Using the spGRN
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Su, J.; Song, Y.; Zhu, Z.; Huang, X.; Fan, J.; Qiao, J.; Mao, F. Cell-cell communication: New insights and clinical implications. Signal Transduct. Target. Ther. 2024, 9, 196. [Google Scholar] [CrossRef]
- Schürch, C.M.; Bhate, S.S.; Barlow, G.L.; Phillips, D.J.; Noti, L.; Zlobec, I.; Chu, P.; Black, S.; Demeter, J.; McIlwain, D.R.; et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 2020, 182, 1341–1359.e1319. [Google Scholar] [CrossRef]
- Wang, S.; Sun, S.T.; Zhang, X.Y.; Ding, H.R.; Yuan, Y.; He, J.J.; Wang, M.S.; Yang, B.; Li, Y.B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int. J. Mol. Sci. 2023, 24, 2943. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Lu, X.; Shao, X.; Zhu, L.; Fan, X. Uncovering an Organ’s Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. Trends Biotechnol. 2021, 39, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.G.; Chávez-Fuentes, J.C.; O’Brien, M.; Xu, J.; Ruiz, E.; Wang, W.; Amin, I.; Sarfraz, I.; Guckhool, P.; Sistig, A.; et al. Giotto Suite: A multi-scale and technology-agnostic spatial multi-omics analysis ecosystem. arXiv 2023, arXiv:2023.11.26.568752. [Google Scholar]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, W.; Jin, H.; Chen, X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell-Cell Communication. Cells 2023, 12, 1970. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, S.; Liu, Z.; Xu, L.; Chen, L.; Xie, L. Single-Cell RNA Sequencing Analysis of Gene Regulatory Network Changes in the Development of Lung Adenocarcinoma. Biomolecules 2023, 13, 671. [Google Scholar] [CrossRef]
- Xu, K.; Yu, D.; Zhang, S.; Chen, L.; Liu, Z.; Xie, L. Deciphering the Immune Microenvironment at the Forefront of Tumor Aggressiveness by Constructing a Regulatory Network with Single-Cell and Spatial Transcriptomic Data. Genes 2024, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Baghdassarian, H.M.; Lewis, N.E. The diversification of methods for studying cell-cell interactions and communication. Nat. Rev. Genet. 2024, 25, 381–400. [Google Scholar] [CrossRef]
- Peng, L.; Wang, F.; Wang, Z.; Tan, J.; Huang, L.; Tian, X.; Liu, G.; Zhou, L. Cell-cell communication inference and analysis in the tumour microenvironments from single-cell transcriptomics: Data resources and computational strategies. Brief Bioinform. 2022, 23, bbac234. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ma, W.; Zang, Y.; Guo, Y.; Li, Y.; Zhang, Y.; Dong, X.; Liu, Y.; Zhan, X.; Pan, Z.; et al. Spatially organized tumor-stroma boundary determines the efficacy of immunotherapy in colorectal cancer patients. Nat. Commun. 2024, 15, 10259. [Google Scholar] [CrossRef]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Yuan, Y.; Zhu, Y. What is new in cancer-associated fibroblast biomarkers? Cell Commun. Signal. 2023, 21, 96. [Google Scholar] [CrossRef]
- Xia, J.; Xie, Z.; Niu, G.; Lu, Z.; Wang, Z.; Xing, Y.; Ren, J.; Hu, Z.; Hong, R.; Cao, Z.; et al. Single-cell landscape and clinical outcomes of infiltrating B cells in colorectal cancer. Immunology 2023, 168, 135–151. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, G.; Ren, X.; Yao, Z.; Zhou, Q.; Ren, X.; Chen, S.; Xu, L.; Sun, K.; Zeng, Q.; et al. Cross-talk between Myeloid and B Cells Shapes the Distinct Microenvironments of Primary and Secondary Liver Cancer. Cancer Res. 2023, 83, 3544–3561. [Google Scholar] [CrossRef]
- Mani, D.R.; Krug, K.; Zhang, B.; Satpathy, S.; Clauser, K.R.; Ding, L.; Ellis, M.; Gillette, M.A.; Carr, S.A. Cancer proteogenomics: Current impact and future prospects. Nat. Rev. Cancer 2022, 22, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Steen, J.A.; Mann, M. Mass-spectrometry-based proteomics: From single cells to clinical applications. Nature 2025, 638, 901–911. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e338. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Zheng, X.; Song, J.; Yu, C.; Zhou, Z.; Liu, X.; Yu, J.; Xu, G.; Yang, J.; He, X.; Bai, X.; et al. Single-cell transcriptomic profiling unravels the adenoma-initiation role of protein tyrosine kinases during colorectal tumorigenesis. Signal Transduct. Target. Ther. 2022, 7, 60. [Google Scholar] [CrossRef]
- Hua, Y.; Ma, X.; Zhao, X.; Wei, X.; Mu, X.; Zhang, X. Characterization of metastasis-specific macrophages in colorectal cancer for prognosis prediction and immunometabolic remodeling. Sci. Rep. 2024, 14, 26361. [Google Scholar] [CrossRef]
- Du, J.; Zhang, J.; Wang, L.; Wang, X.; Zhao, Y.; Lu, J.; Fan, T.; Niu, M.; Zhang, J.; Cheng, F.; et al. Selective oxidative protection leads to tissue topological changes orchestrated by macrophage during ulcerative colitis. Nat. Commun. 2023, 14, 3675. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.; Lou, J.; Wu, L.; Zhang, S.; Liu, X.; Ke, Y.; Zhao, S.; Song, Z.; Bai, X.; et al. Machine Learning Integration with Single-Cell Transcriptome Sequencing Datasets Reveals the Impact of Tumor-Associated Neutrophils on the Immune Microenvironment and Immunotherapy Outcomes in Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 12715. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Chen, Y.; Pal, B.; Lindeman, G.J.; Visvader, J.E.; Smyth, G.K. R code and downstream analysis objects for the scRNA-seq atlas of normal and tumorigenic human breast tissue. Sci. Data 2022, 9, 96. [Google Scholar] [CrossRef]
- Parsons, A.; Colon, E.S.; Spasic, M.; Kurt, B.B.; Swarbrick, A.; Freedman, R.A.; Mittendorf, E.A.; van Galen, P.; McAllister, S.S. Cell Populations in Human Breast Cancers are Molecularly and Biologically Distinct with Age. arXiv 2024, arXiv:rs.3.rs-5167339. [Google Scholar]
- Xu, J.; Fang, Y.; Chen, K.; Li, S.; Tang, S.; Ren, Y.; Cen, Y.; Fei, W.; Zhang, B.; Shen, Y.; et al. Single-Cell RNA Sequencing Reveals the Tissue Architecture in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2022, 28, 3590–3602. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Huang, Z.; Wu, C.; Zhang, F.; Han, A.; Stalin, A.; Lu, S.; Guo, S.; Huang, J.; et al. T cell-related prognostic risk model and tumor immune environment modulation in lung adenocarcinoma based on single-cell and bulk RNA sequencing. Comput. Biol. Med. 2023, 152, 106460. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef]
- Xun, Z.; Ding, X.; Zhang, Y.; Zhang, B.; Lai, S.; Zou, D.; Zheng, J.; Chen, G.; Su, B.; Han, L.; et al. Reconstruction of the tumor spatial microenvironment along the malignant-boundary-nonmalignant axis. Nat. Commun. 2023, 14, 933. [Google Scholar] [CrossRef]
- Shao, X.; Li, C.; Yang, H.; Lu, X.; Liao, J.; Qian, J.; Wang, K.; Cheng, J.; Yang, P.; Chen, H.; et al. Knowledge-graph-based cell-cell communication inference for spatially resolved transcriptomic data with SpaTalk. Nat. Commun. 2022, 13, 4429. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.; Tan, X.; Balderson, B.; Xu, J.; Grice, L.F.; Yoon, S.; Willis, E.F.; Tran, M.; Lam, P.Y.; Raghubar, A.; et al. Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues. Nat. Commun. 2023, 14, 7739. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef]
- Xie, L.; Kong, Q.; Ai, M.; He, A.; Yao, B.; Zhang, L.; Zhang, K.; Zhu, C.; Li, Y.; Xia, L.; et al. Spatial Proteomic Profiling of Colorectal Cancer Revealed Its Tumor Microenvironment Heterogeneity. J. Proteome. Res. 2024, 23, 3342–3352. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Quail, D.F. Decoding the tumor microenvironment with spatial technologies. Nat. Immunol. 2023, 24, 1982–1993. [Google Scholar] [CrossRef]
- Sternberg, C.; Raigel, M.; Limberger, T.; Trachtová, K.; Schlederer, M.; Lindner, D.; Kodajova, P.; Yang, J.; Ziegler, R.; Kalla, J.; et al. Cell-autonomous IL6ST activation suppresses prostate cancer development via STAT3/ARF/p53-driven senescence and confers an immune-active tumor microenvironment. Mol. Cancer 2024, 23, 245. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Kim, D.H.; Sung, M.; Park, M.S.; Sun, E.G.; Yoon, S.; Yoo, K.H.; Radhakrishnan, K.; Jung, S.Y.; Bae, W.K.; Cho, S.H.; et al. Galectin 3-binding protein (LGALS3BP) depletion attenuates hepatic fibrosis by reducing transforming growth factor-β1 (TGF-β1) availability and inhibits hepatocarcinogenesis. Cancer Commun. 2024, 44, 1106–1129. [Google Scholar] [CrossRef]
- Hou, C.; Wang, D.; Zhao, M.; Ballar, P.; Zhang, X.; Mei, Q.; Wang, W.; Li, X.; Sheng, Q.; Liu, J.; et al. MANF brakes TLR4 signaling by competitively binding S100A8 with S100A9 to regulate macrophage phenotypes in hepatic fibrosis. Acta Pharm. Sin. B 2023, 13, 4234–4252. [Google Scholar] [CrossRef] [PubMed]
- Torretta, S.; Scagliola, A.; Ricci, L.; Mainini, F.; Di Marco, S.; Cuccovillo, I.; Kajaste-Rudnitski, A.; Sumpton, D.; Ryan, K.M.; Cardaci, S. D-mannose suppresses macrophage IL-1β production. Nat. Commun. 2020, 11, 6343. [Google Scholar] [CrossRef]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A.; et al. Loss of HIF1A From Pancreatic Cancer Cells Increases Expression of PPP1R1B and Degradation of p53 to Promote Invasion and Metastasis. Gastroenterology 2020, 159, 1882–1897.e1885. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gutiérrez, L.; Ferrara, N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat. Rev. Mol. Cell. Biol. 2023, 24, 816–834. [Google Scholar] [CrossRef]
- Lee, C.; Chen, R.; Sun, G.; Liu, X.; Lin, X.; He, C.; Xing, L.; Liu, L.; Jensen, L.D.; Kumar, A.; et al. VEGF-B prevents excessive angiogenesis by inhibiting FGF2/FGFR1 pathway. Signal Transduct. Target. Ther. 2023, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimnezhad, M.; Valizadeh, A.; Majidinia, M.; Tabnak, P.; Yousefi, B. Unveiling the potential of FOXO3 in lung cancer: From molecular insights to therapeutic prospects. Biomed. Pharmacother. 2024, 176, 116833. [Google Scholar] [CrossRef]
- Tsao, A.S.; Wei, W.; Kuhn, E.; Spencer, L.; Solis, L.M.; Suraokar, M.; Lee, J.J.; Hong, W.K.; Wistuba, I.I. Immunohistochemical overexpression of platelet-derived growth factor receptor-beta (PDGFR-β) is associated with PDGFRB gene copy number gain in sarcomatoid non-small-cell lung cancer. Clin. Lung Cancer 2011, 12, 369–374. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Li, J.; Shu, X.; Xu, J.; Su, S.M.; Chan, U.I.; Mo, L.; Liu, J.; Zhang, X.; Adhav, R.; Chen, Q.; et al. S100A9-CXCL12 activation in BRCA1-mutant breast cancer promotes an immunosuppressive microenvironment associated with resistance to immunotherapy. Nat. Commun. 2022, 13, 1481. [Google Scholar] [CrossRef]
- Davidson, B.; Stavnes, H.T.; Holth, A.; Chen, X.; Yang, Y.; Shih Ie, M.; Wang, T.L. Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from breast carcinoma in effusions. J. Cell. Mol. Med. 2011, 15, 535–544. [Google Scholar] [CrossRef]
- Tian, X.; Song, J.; Zhang, X.; Yan, M.; Wang, S.; Wang, Y.; Xu, L.; Zhao, L.; Wei, J.J.; Shao, C.; et al. MYC-regulated pseudogene HMGA1P6 promotes ovarian cancer malignancy via augmenting the oncogenic HMGA1/2. Cell Death Dis. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tao, X.; Chen, P.; Jiang, P.; Li, W.; Chang, H.; Wei, C.; Lai, X.; Zhang, H.; Pan, Y.; et al. MEK inhibition prevents CAR-T cell exhaustion and differentiation via downregulation of c-Fos and JunB. Signal Transduct. Target. Ther. 2024, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Zhou, Z.; Ma, Z.; Li, Z.; Liu, C.; Huang, S.; Zhang, C.; Hou, B. Characterization of the prognostic and oncologic values of ITGB superfamily members in pancreatic cancer. J. Cell. Mol. Med. 2020, 24, 13481–13493. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Almet, A.A.; Nie, Q. The promising application of cell-cell interaction analysis in cancer from single-cell and spatial transcriptomics. Semin. Cancer Biol. 2023, 95, 42–51. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, B.; Ming, W.; Zhou, X.; Wang, J.; Chen, D. SpaTopic: A statistical learning framework for exploring tumor spatial architecture from spatially resolved transcriptomic data. Sci. Adv. 2024, 10, eadp4942. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Xu, K.; Zhang, S.; Chen, L.; Liu, Z.; Xie, L. Construction of Gene Regulatory Networks Based on Spatial Multi-Omics Data and Application in Tumor-Boundary Analysis. Genes 2025, 16, 821. https://doi.org/10.3390/genes16070821
Du Y, Xu K, Zhang S, Chen L, Liu Z, Xie L. Construction of Gene Regulatory Networks Based on Spatial Multi-Omics Data and Application in Tumor-Boundary Analysis. Genes. 2025; 16(7):821. https://doi.org/10.3390/genes16070821
Chicago/Turabian StyleDu, Yiwen, Kun Xu, Siwen Zhang, Lanming Chen, Zhenhao Liu, and Lu Xie. 2025. "Construction of Gene Regulatory Networks Based on Spatial Multi-Omics Data and Application in Tumor-Boundary Analysis" Genes 16, no. 7: 821. https://doi.org/10.3390/genes16070821
APA StyleDu, Y., Xu, K., Zhang, S., Chen, L., Liu, Z., & Xie, L. (2025). Construction of Gene Regulatory Networks Based on Spatial Multi-Omics Data and Application in Tumor-Boundary Analysis. Genes, 16(7), 821. https://doi.org/10.3390/genes16070821