
Cytogenetics and Cytogenomics in Clinical Diagnostics: Genome Architecture, Structural Variants, and Translational Applications

 , , , and
, , , and 
Abstract

1. Introduction
2. The Nuclear Genome Architecture in Health and Disease
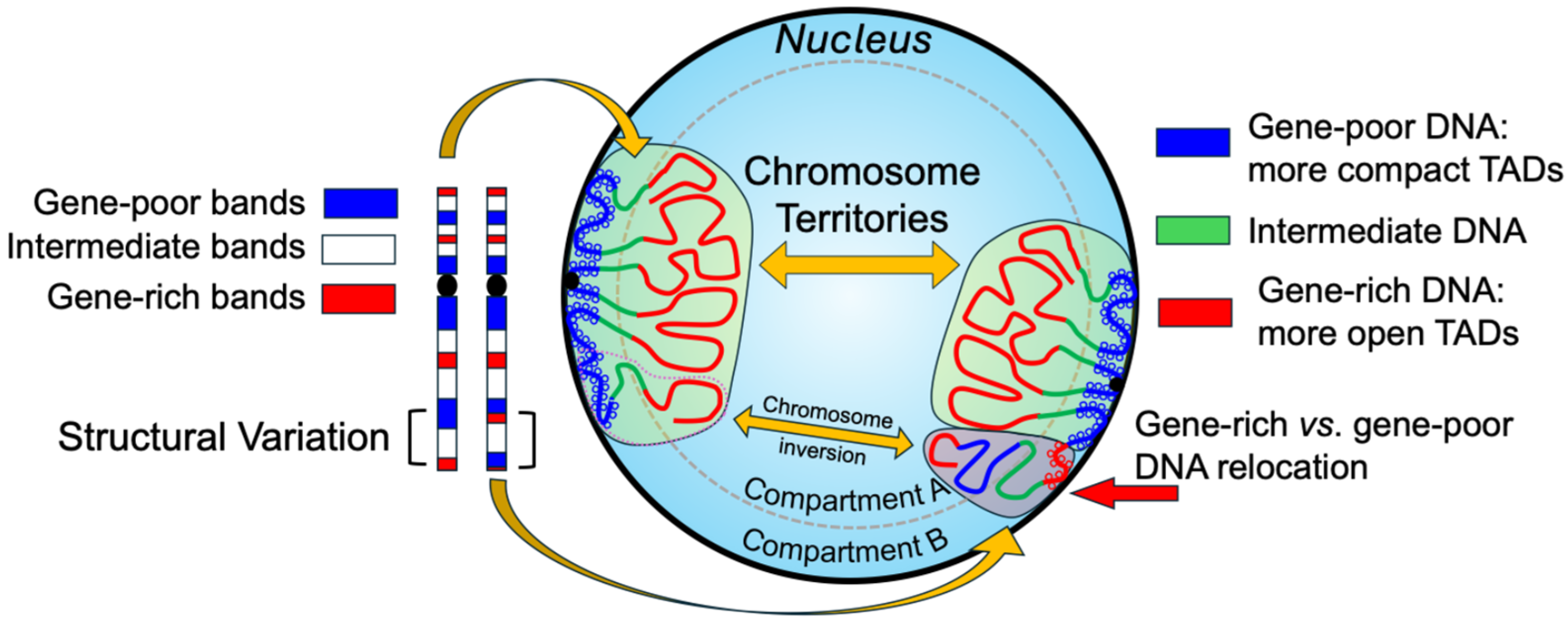
2.1. Spatial Organization of the Genome and Chromosome Territories
2.2. Topologically Associating Domains and 3D Gene Regulation
2.3. Nuclear Architecture Alterations in Genetic and Neoplastic Disorders
2.4. Diagnostic Implications and Future Perspectives of Functional Cytogenomics
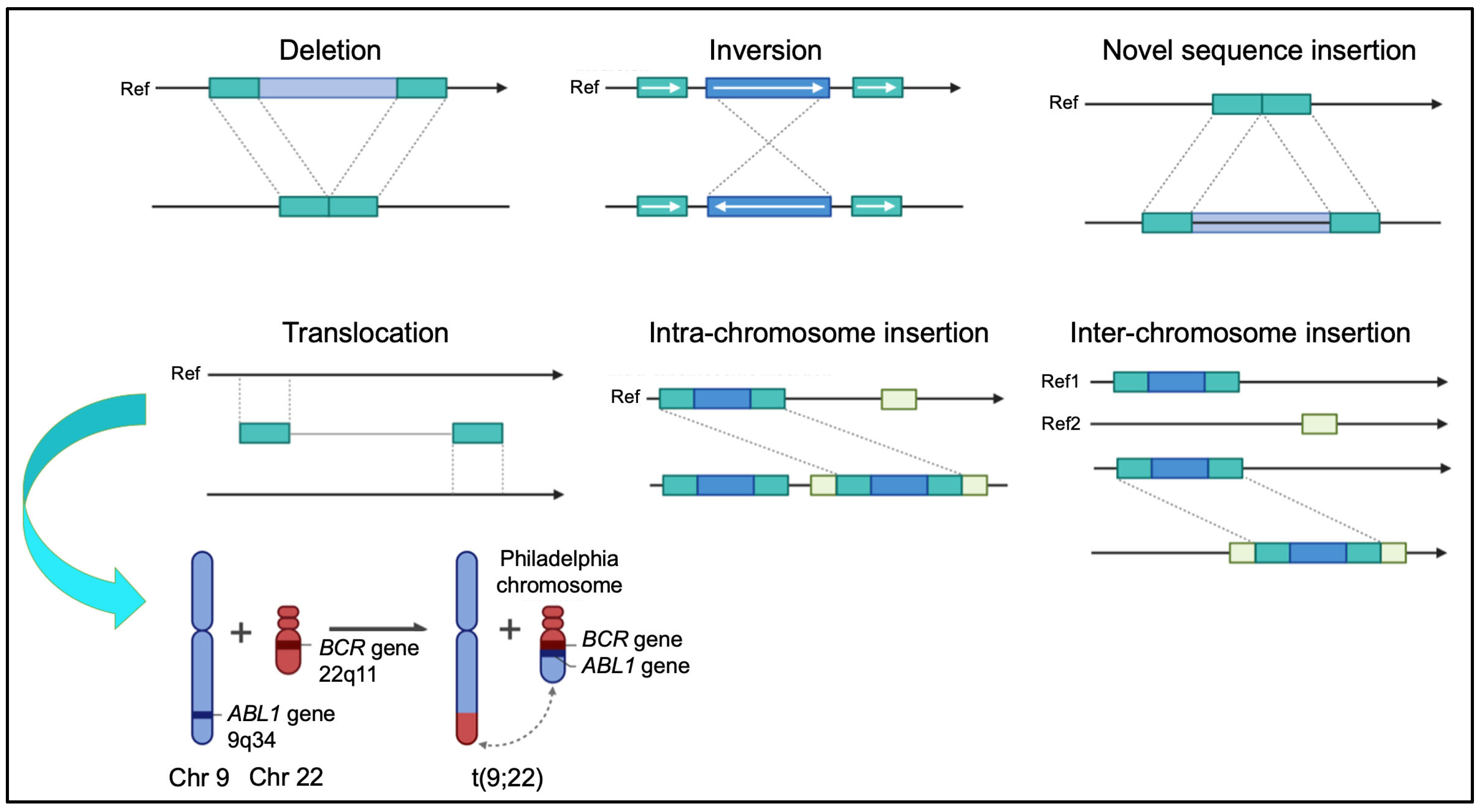
3. Chromosomal Rearrangements: Mechanisms and Functional Implications
3.1. Mechanisms of Formation
- Non-Homologous End Joining (NHEJ): Repairs double-strand breaks without a homologous template, often generating small insertions or deletions and facilitating both balanced and unbalanced translocations. This mechanism is commonly implicated in lymphoid malignancies, where antigen receptor gene rearrangements misfire [39].
- Fork Stalling and Template Switching (FoSTeS): Occurs during DNA replication when stalled forks switch templates, resulting in complex rearrangements such as deletions, duplications, and copy number variants [40].
- Chromothripsis: A catastrophic event causing chromosome fragmentation and erroneous reassembly. This process can generate tens to hundreds of clustered rearrangements and is increasingly recognized as a driver of aggressive cancers such as glioblastoma, osteosarcoma, and some hematologic malignancies [41].
- Chromoanasynthesis: A replication-based mechanism involving serial template switching during DNA synthesis, giving rise to highly complex SVs with duplications, triplications, and microhomology at breakpoint junctions. It is frequently associated with congenital malformation syndromes and neurodevelopmental disorders [42].
- Chromoplexy: A process involving coordinated, interdependent DNA strand breaks and re-ligation events across multiple chromosomes. It generates complex but often balanced rearrangements and is particularly enriched in prostate and other epithelial cancers, where it can simultaneously affect multiple oncogenes and tumor suppressors [43].
3.2. Functional Impact
- Gene Disruption and Regulatory Alterations: SVs may interrupt coding sequences or reposition genes relative to regulatory elements (e.g., enhancers). Such disruptions may lead to a loss of gene function, inappropriate expression, or dosage imbalances [47]. For example, duplications upstream of the IHH gene cause ectopic enhancer interactions leading to digit malformations such as brachydactyly or syndactyl [48].
- TAD Disruption: SVs can break or fuse TAD boundaries, altering chromatin loops and long-range gene regulation. SVs such as deletions, duplications, and inversions can fuse or separate TADs, alter chromatin loop dynamics, and facilitate inappropriate enhancer–promoter contacts. This “enhancer hijacking” mechanism can lead to tissue-specific gene misexpression, underlying some developmental disorders and cancers, as seen in duplications near the EPHA4 locus that cause limb abnormalities [49]. In cancers, TAD disruptions may activate proto-oncogenes or silence tumor suppressors through similar architectural perturbations.
3.3. Clinically Relevant Rearrangements
- Balanced Translocations: Balanced chromosomal translocations often result in gene fusions with oncogenic potential. A classical example is the BCR::ABL1 fusion in chronic myeloid leukemia (CML) [50]. Similarly, the t(15;17) translocation in acute promyelocytic leukemia (APL) produces the PML::RARA fusion, rendering the disease highly responsive to differentiation therapy with all-trans retinoic acid (ATRA) and arsenic trioxide [51].
- Interstitial Deletions: Deletions on chromosome arms, particularly on 5q and 20q, are frequent in myeloid malignancies such as myelodysplastic syndromes (MDS), often associated with a favorable prognosis by disrupting tumor suppressor genes or regulatory regions [52]. In contrast, deletions affecting 7q and 17p correlate with aggressive disease and poor outcomes [53].
- Complex SVs and Chromothripsis: Chromothripsis causes massive localized chromosomal rearrangements that drive oncogenesis by forming oncogenic fusions or deleting tumor suppressors. It is observed in hematologic malignancies and solid tumors like neuroblastoma and glioblastoma, which are frequently linked to treatment resistance and a poorer prognosis [54,55].
- Constitutional Rearrangements: Microdeletions and duplications cause syndromes such as 22q11.2 deletion syndrome (DiGeorge syndrome), affecting the cardiac, immune, and neurodevelopmental systems [52]. Similarly, microdeletions/duplications at 15q13.2–q13.3 and cryptic chromosomal aberrations contribute to autism spectrum disorder and intellectual disability [56,57]. These rearrangements are often cryptic and may be missed by conventional cytogenetic techniques.
4. Replication Timing and Chromosome Banding in Cytogenomic Diagnostics
4.1. Replication Timing and Nuclear Architecture
4.2. Replication Timing as a Functional Marker
4.3. Epigenetic Regulation and Genome Stability
4.4. Chromosomal Banding and Epigenetic Organization
4.5. Replication Stress and Disease Mechanisms
4.6. Chromosomal Rearrangements in Human Pathologies
5. Diagnostic Technologies: From Conventional Cytogenetics to High-Resolution Genomics
5.1. Classical Cytogenetics: Principles, Applications, and Limitations
5.2. Fluorescence In Situ Hybridization: Increased Specificity and Sensitivity
5.3. High-Resolution Genomic Technologies: Array-CGH, SNP Arrays, and Optical Mapping
5.4. Genomic Sequencing: Short-Read and Long-Read
5.5. Current Limitations and Clinical Applicability of Cytogenomic Technologies
6. Cytogenomics in Cancer Diagnosis and Prognosis
6.1. Chromosomal Rearrangements in Cancer Diagnosis and Prognosis
6.2. Advances in Cytogenomic Technologies and Their Clinical Impact
7. Integrating 3D Genome and Epigenome Data in Clinical Contexts
7.1. Emerging Techniques for 3D Genome and Epigenomic Profiling
7.2. Examples of Clinical Applications
7.3. Towards Integrated Functional Cytogenomics
7.4. Future Perspectives
8. Challenges and Future Perspectives
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3C | Chromosome Conformation Capture |
| 3D | Three-dimensional |
| 4C | Circular Chromosome Conformation Capture |
| 5C | Carbon Copy Chromosome Conformation Capture |
| AI | Artificial Intelligence |
| ALL | Acute Lymphoblastic Leukemia |
| AML | Acute Myeloid Leukemia |
| APL | Acute Promyelocytic Leukemia |
| ATAC-seq | Assay for Transposase-Accessible Chromatin using sequencing |
| ATRA | All-Trans Retinoic Acid |
| B-ALL | B-cell Acute Lymphoblastic Leukemia |
| CGH | Comparative Genomic Hybridization |
| ChIP-seq | Chromatin Immunoprecipitation sequencing |
| CLL | Chronic Lymphocytic Leukemia |
| CMA | Chromosomal Microarrays |
| CML | Chronic Myeloid Leukemia |
| CNS | Central Nervous System |
| CNV | Copy Number Variant |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMR | Differentially Methylated Region |
| ESC | Embryonic Stem Cell |
| FISH | Fluorescence In Situ Hybridization |
| FoSTeS | Fork Stalling and Template Switching |
| FRET | Förster Resonance Energy Transfer |
| G-banding | Giemsa banding |
| GWAS | Genome-Wide Association Study |
| H&E | Hematoxylin and Eosin |
| H3K27me3 | Histone 3 Lysine 27 trimethylation |
| HDAC | Histone Deacetylase |
| Hi-C | High-throughput Chromosome Conformation Capture |
| ICF syndrome | Immunodeficiency, Centromeric instability, and Facial anomalies syndrome |
| iPSC | Induced Pluripotent Stem Cell |
| LOH | Loss of Heterozygosity |
| MDS | Myelodysplastic Syndromes |
| MMEJ | Microhomology-Mediated End Joining |
| NAHR | Non-Allelic Homologous Recombination |
| NHEJ | Non-Homologous End Joining |
| OGM | Optical Genome Mapping |
| TAD | Topologically Associating Domain |
| TERT | Telomerase Reverse Transcriptase |
| TF | Transcription Factor |
| WES | Whole-Exome Sequencing |
| WGS | Whole-Genome Sequencing |
References
- Balciuniene, J.; Ning, Y.; Lazarus, H.M.; Aikawa, V.; Sherpa, S.; Zhang, Y.; Morrissette, J.J.D. Cancer Cytogenetics in a Genomics World: Wedding the Old with the New. Blood Rev. 2024, 66, 101209. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T. Cytogenetic Contribution to Uniparental Disomy (UPD). Mol. Cytogenet. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome Structural Variation Discovery and Genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the Diverse Spectrum of Inversions, Complex Structural Variation, and Chromothripsis in the Morbid Human Genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef]
- Federico, C.; Bruno, F.; Ragusa, D.; Clements, C.S.; Brancato, D.; Henry, M.P.; Bridger, J.M.; Tosi, S.; Saccone, S. Chromosomal Rearrangements and Altered Nuclear Organization: Recent Mechanistic Models in Cancer. Cancers 2021, 13, 5860. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and Function of the 3D Genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural Variation in the 3D Genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Van Steensel, B.; Furlong, E.E.M. The Role of Transcription in Shaping the Spatial Organization of the Genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 327–337. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D Nucleome: Evidence for a Dynamic Nuclear Landscape Based on Co-aligned Active and Inactive Nuclear Compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef]
- Saccone, S.; Federico, C.; Bernardi, G. Localization of the Gene-Richest and the Gene-Poorest Isochores in the Interphase Nuclei of Mammals and Birds. Gene 2002, 300, 169–178. [Google Scholar] [CrossRef]
- Lanctôt, C.; Cheutin, T.; Cremer, M.; Cavalli, G.; Cremer, T. Dynamic Genome Architecture in the Nuclear Space: Regulation of Gene Expression in Three Dimensions. Nat. Rev. Genet. 2007, 8, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A. The Spatial Organization of the Human Genome. Annu. Rev. Genom. Hum. Genet. 2013, 14, 67–84. [Google Scholar] [CrossRef]
- Maass, P.G.; Barutcu, A.R.; Rinn, J.L. Interchromosomal Interactions: A Genomic Love Story of Kissing Chromosomes. J. Cell Biol. 2019, 218, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Federico, C.; Cantarella, C.D.; Di Mare, P.; Tosi, S.; Saccone, S. The Radial Arrangement of the Human Chromosome 7 in the Lymphocyte Cell Nucleus Is Associated with Chromosomal Band Gene Density. Chromosoma 2008, 117, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Daban, J. Rethinking Models of DNA Organization in Micrometer-Sized Chromosomes from the Perspective of the Nanoproperties of Chromatin Favoring a Multilayer Structure. Small Struct. 2024, 5, 2400203. [Google Scholar] [CrossRef]
- Ye, C.J.; Stilgenbauer, L.; Moy, A.; Liu, G.; Heng, H.H. What Is Karyotype Coding and Why Is Genomic Topology Important for Cancer and Evolution? Front. Genet. 2019, 10, 1082. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Karyotype Coding: The Creation and Maintenance of System Information for Complexity and Biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef]
- Furst, R. The Importance of Henry H. Heng’s Genome Architecture Theory. Prog. Biophys. Mol. Biol. 2021, 165, 153–156. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Naumova, N.; Imakaev, M.; Fudenberg, G.; Zhan, Y.; Lajoie, B.R.; Mirny, L.A.; Dekker, J. Organization of the Mitotic Chromosome. Science 2013, 342, 948–953. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M.; Hübner, B.; Silahtaroglu, A.; Hendzel, M.; Lanctôt, C.; Strickfaden, H.; Cremer, C. The Interchromatin Compartment Participates in the Structural and Functional Organization of the Cell Nucleus. BioEssays 2020, 42, 1900132. [Google Scholar] [CrossRef]
- Ballabio, E.; Cantarella, C.D.; Federico, C.; Di Mare, P.; Hall, G.; Harbott, J.; Hughes, J.; Saccone, S.; Tosi, S. Ectopic Expression of the HLXB9 Gene Is Associated with an Altered Nuclear Position in t(7;12) Leukaemias. Leukemia 2009, 23, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Federico, C.; Pappalardo, A.M.; Ferrito, V.; Tosi, S.; Saccone, S. Genomic Properties of Chromosomal Bands Are Linked to Evolutionary Rearrangements and New Centromere Formation in Primates. Chromosome Res. 2017, 25, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Federico, C.; Brancato, D.; Bruno, F.; Galvano, D.; Caruso, M.; Saccone, S. Robertsonian Translocation between Human Chromosomes 21 and 22, Inherited across Three Generations, without Any Phenotypic Effect. Genes 2024, 15, 722. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C Deficiency Causes Defective Nuclear Mechanics and Mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef]
- Bridger, J.M.; Foeger, N.; Kill, I.R.; Herrmann, H. The Nuclear Lamina: Both a Structural Framework and a Platform for Genome Organization. FEBS J. 2007, 274, 1354–1361. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The Role of Nuclear Lamin B1 in Cell Proliferation and Senescence. Genes. Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Erdos, M.R.; Ried, T. The Role of Lamin B1 for the Maintenance of Nuclear Structure and Function. Nucleus 2015, 6, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Bird, A. Rett Syndrome: A Complex Disorder with Simple Roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef]
- Brancato, D.; Bruno, F.; Coniglio, E.; Sturiale, V.; Saccone, S.; Federico, C. The Chromatin Organization Close to SNP Rs12913832, Involved in Eye Color Variation, Is Evolutionary Conserved in Vertebrates. Int. J. Mol. Sci. 2024, 25, 6602. [Google Scholar] [CrossRef]
- Visser, M.; Kayser, M.; Grosveld, F.; Palstra, R. Genetic Variation in Regulatory DNA Elements: The Case of OCA 2 Transcriptional Regulation. Pigment. Cell Melanoma Res. 2014, 27, 169–177. [Google Scholar] [CrossRef]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef]
- Deng, S.; Feng, Y.; Pauklin, S. 3D Chromatin Architecture and Transcription Regulation in Cancer. J. Hematol. Oncol. 2022, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural Variation in the Human Genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Human Nonhomologous DNA End Joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy Number Variation in Human Health, Disease, and Evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Kleinfinger, P.; Cances, C.; Bazin, A.; Julia, S.; Trost, D.; Lohmann, L.; Vigouroux, A. Constitutional Chromoanasynthesis: Description of a Rare Chromosomal Event in a Patient. Eur. J. Med. Genet. 2014, 57, 567–570. [Google Scholar] [CrossRef]
- Pellestor, F.; Ganne, B.; Gaillard, J.B.; Gatinois, V. Chromoplexy: A Pathway to Genomic Complexity and Cancer Development. Int. J. Mol. Sci. 2025, 26, 3826. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Malkova, A. Break Induced Replication in Eukaryotes: Mechanisms, Functions, and Consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural Variation in the Human Genome and Its Role in Disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H. Roles for Retrotransposon Insertions in Human Disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic Impact of Genomic Structural Variation: Insights from and for Human Disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef]
- Botten, G.; Zhang, Y.; Dudnyk, K.; Kim, Y.J.; Liu, X.; Sanders, J.T.; Imanci, A.; Droin, N.M.; Cao, H.; Kaphle, P.; et al. Structural Variation Cooperates with Permissive Chromatin to Control Enhancer Hijacking-Mediated Oncogenic Transcription. Blood 2023, 142, 336–351. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016, 32, 225–237. [Google Scholar] [CrossRef]
- Hehlmann, R.; Hochhaus, A.; Baccarani, M. Chronic Myeloid Leukaemia. Lancet 2007, 370, 342–350. [Google Scholar] [CrossRef]
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug Therapy for Acute Myeloid Leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive Analysis of Chromothripsis in 2,658 Human Cancers Using Whole-Genome Sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Van Sluis, P.; Valentijn, L.J.; Van Der Ploeg, I.; Hamdi, M.; Van Nes, J.; Westerman, B.A.; Van Arkel, J.; et al. Sequencing of Neuroblastoma Identifies Chromothripsis and Defects in Neuritogenesis Genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Brand, H.; Pillalamarri, V.; Collins, R.L.; Eggert, S.; O’Dushlaine, C.; Braaten, E.B.; Stone, M.R.; Chambert, K.; Doty, N.D.; Hanscom, C.; et al. Cryptic and Complex Chromosomal Aberrations in Early-Onset Neuropsychiatric Disorders. Am. J. Hum. Genet. 2014, 95, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J.; et al. Microdeletion/Duplication at 15q13.2q13.3 among Individuals with Features of Autism and Other Neuropsychiatric Disorders. J. Med. Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; Von Haeseler, A.; Schatz, M.C. Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Sima, J.; Gilbert, D.M. Complex Correlations: Replication Timing and Mutational Landscapes during Cancer and Genome Evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication Timing and Transcriptional Control: Beyond Cause and Effect—Part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA Replication Timing in the 3D Genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zheng, H.; Huang, B.; Ma, R.; Wu, J.; Zhang, X.; He, J.; Xiang, Y.; Wang, Q.; Li, Y.; et al. Allelic Reprogramming of 3D Chromatin Architecture during Early Mammalian Development. Nature 2017, 547, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically Associating Domains Are Stable Units of Replication-Timing Regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Federico, C.; Saccone, S.; Bernardi, G. The Gene-Richest Bands of Human Chromosomes Replicate at the Onset of the S-Phase. Cytogenet. Genome Res. 1998, 80, 83–88. [Google Scholar] [CrossRef]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily Conserved Replication Timing Profiles Predict Long-Range Chromatin Interactions and Distinguish Closely Related Cell Types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.J.; Shore, D. Rif1 Controls DNA Replication Timing in Yeast through the PP1 Phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global Reorganization of Replication Domains During Embryonic Stem Cell Differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.-B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; Van Steensel, B. Constitutive Nuclear Lamina–Genome Interactions Are Highly Conserved and Associated with A/T-Rich Sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential Relationship of DNA Replication Timing to Different Forms of Human Mutation and Variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Dutrillaux, B.; Couturier, J.; Richer, C.-L.; Viegas-Péquignot, E. Sequence of DNA Replication in 277 R- and Q-Bands of Human Chromosomes Using a BrdU Treatment. Chromosoma 1976, 58, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Bernardi, G. Replication Timing, Chromosomal Bands, and Isochores. Proc. Natl. Acad. Sci. USA 2008, 105, 3433–3437. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. Structural and Evolutionary Genomics: Natural Selection in Genome Evolution; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 978-1-281-22736-2. [Google Scholar]
- Saccone, S.; Pavliček, A.; Federico, C.; Pačes, J.; Bernardi, G. Genes, Isochores and Bands in Human Chromosomes 21 and 22. Chromosome Res. 2001, 9, 533–539. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium; Whitehead Institute for Biomedical Research, Center for Genome Research; Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, C. Chromosome Territories, Nuclear Architecture and Gene Regulation in Mammalian Cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-Type-Specific Replication Initiation Programs Set Fragility of the FRA3B Fragile Site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome Fragile Sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Weterings, E.; Chen, D.J. The Endless Tale of Non-Homologous End-Joining. Cell Res. 2008, 18, 114–124. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA Repair by Nonhomologous End Joining and Homologous Recombination during Cell Cycle in Human Cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium; Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; et al. The Impact of Structural Variation on Human Gene Expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef]
- Liehr, T. Molecular Cytogenetics in the Era of Chromosomics and Cytogenomic Approaches. Front. Genet. 2021, 12, 720507. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Lupski, J.R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 2000, 34, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J. High Resolution of Human Chromosomes. Science 1976, 191, 1268–1270. [Google Scholar] [CrossRef]
- Lichter, P.; Tang, C.-J.C.; Call, K.; Hermanson, G.; Evans, G.A.; Housman, D.; Ward, D.C. High-Resolution Mapping of Human Chromosome 11 by in Situ Hybridization with Cosmid Clones. Science 1990, 247, 64–69. [Google Scholar] [CrossRef]
- Trask, B. Fluorescence in Situ Hybridization: Applications in Cytogenetics and Gene Mapping. Trends Genet. 1991, 7, 149–154. [Google Scholar] [CrossRef]
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of Individual Human Chromosomes in Metaphase and Interphase Cells by in Situ Suppression Hybridization Using Recombinant DNA Libraries. Hum. Genet. 1988, 80, 224–234. [Google Scholar] [CrossRef]
- Lawrence, J. Sensitive, High-Resolution Chromatin and Chromosome Mapping in Situ: Presence and Orientation of Two Closely Integrated Copies of EBV in a Lymphoma Line. Cell 1988, 52, 51–61. [Google Scholar] [CrossRef]
- Schröck, E.; Du Manoir, S.; Veldman, T.; Schoell, B.; Wienberg, J.; Ferguson-Smith, M.A.; Ning, Y.; Ledbetter, D.H.; Bar-Am, I.; Soenksen, D.; et al. Multicolor Spectral Karyotyping of Human Chromosomes. Science 1996, 273, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Raap, A.K.; Florijn, R.J.; Blonden, L.A.J.; Wiegant, J.; Vaandrager, J.-W.; Vrolijk, H.; Den Dunnen, J.; Tanke, H.J.; Van Ommen, G.-J. Fiber FISH as a DNA Mapping Tool. Methods 1996, 9, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Wiegant, J.; Kalle, W.; Mullenders, L.; Brookes, S.; Hoovers, J.M.N.; Dauwerse, J.G.; Van Ommen, G.J.B.; Raap, A.K. High-Resolution in Situ Hybridization Using DNA Halo Preparations. Hum. Mol. Genet. 1992, 1, 587–591. [Google Scholar] [CrossRef]
- Heng, H.H.; Squire, J.; Tsui, L.C. High-Resolution Mapping of Mammalian Genes by in Situ Hybridization to Free Chromatin. Proc. Natl. Acad. Sci. USA 1992, 89, 9509–9513. [Google Scholar] [CrossRef]
- Speicher, M.R.; Ballard, S.G.; Ward, D.C. Karyotyping Human Chromosomes by Combinatorial Multi-Fluor FISH. Nat. Genet. 1996, 12, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Coe, B.P.; Girirajan, S.; Eichler, E.E. The Genetic Variability and Commonality of Neurodevelopmental Disease. Am. J. Med. Genet. Pt. C 2012, 160C, 118–129. [Google Scholar] [CrossRef]
- Gai, X.; Xie, H.M.; Perin, J.C.; Takahashi, N.; Murphy, K.; Wenocur, A.S.; D’arcy, M.; O’Hara, R.J.; Goldmuntz, E.; Grice, D.E.; et al. Rare Structural Variation of Synapse and Neurotransmission Genes in Autism. Mol. Psychiatry 2012, 17, 402–411. [Google Scholar] [CrossRef]
- Spielmann, M.; Mundlos, S. Structural Variations, the Regulatory Landscape of the Genome and Their Alteration in Human Disease. BioEssays 2013, 35, 533–543. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; Van Der Zande, G.; Kater-Baats, E.; Baatout, I.; Van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical Genome Mapping Enables Constitutional Chromosomal Aberration Detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- de Coster, W.; Weissensteiner, M.H.; Sedlazeck, F.J. Towards Population-Scale Long-Read Sequencing. Nat. Rev. Genet. 2021, 22, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Wall, B.P.G.; Nguyen, M.; Harrell, J.C.; Dozmorov, M.G. Machine and deep learning methods for predicting 3D genome organization. In Computational Methods for 3D Genome Analysis; Nakato, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2025; Volume 2856, pp. 357–400. ISBN 978-1-0716-4135-4. [Google Scholar]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Oehler, J.B.; Wright, H.; Stark, Z.; Mallett, A.J.; Schmitz, U. The Application of Long-Read Sequencing in Clinical Settings. Hum. Genom. 2023, 17, 73. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2022. Available online: https://mitelmandatabase.isb-cgc.org/ (accessed on 29 May 2025).
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Bercier, P.; de Thé, H. History of Developing Acute Promyelocytic Leukemia Treatment and Role of Promyelocytic Leukemia Bodies. Cancers 2024, 16, 1351. [Google Scholar] [CrossRef]
- Mullighan, C.G. Molecular Genetics of B-Precursor Acute Lymphoblastic Leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef]
- Melo, J.V.; Barnes, D.J. Chronic Myeloid Leukaemia as a Model of Disease Evolution in Human Cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia Identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Garimberti, E.; Federico, C.; Ragusa, D.; Bruno, F.; Saccone, S.; Bridger, J.M.; Tosi, S. Alterations in Genome Organization in Lymphoma Cell Nuclei Due to the Presence of the t(14;18) Translocation. Int. J. Mol. Sci. 2024, 25, 2377. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- Syndrome Gene by RNA Interference Screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Yu, J.; Qi, Z. Acute Myeloid Leukemia with Inv(16)(P13.1q22) and Deletion of the 5’MYH11/3’CBFB Gene Fusion: A Report of Two Cases and Literature Review. Mol. Cytogenet. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit From Procarbazine, Lomustine, and Vincristine in Oligodendroglial Tumors Is Associated with Mutation of IDH. J. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- The ICGC/TCGAPan-Cancer Analysis of Whole Genomes Consortium; Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; Ahn, K.S.; Ahn, S.-M.; Aikata, H.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational Principles of 3D Genome Architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased Analysis of Potential Targets of Breast Cancer Susceptibility Loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Emerson, D.J.; Zhao, P.A.; Cook, A.L.; Barnett, R.J.; Klein, K.N.; Saulebekova, D.; Ge, C.; Zhou, L.; Simandi, Z.; Minsk, M.K.; et al. Cohesin-Mediated Loop Anchors Confine the Locations of Human Replication Origins. Nature 2022, 606, 812–819. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- The 4D Nucleome Network; Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; et al. The 4D Nucleome Project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin Architecture Reorganization during Stem Cell Differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, X.; Luo, X.; Liu, B.; Tang, J.; Xiao, F.; Wang, W.; Tang, Y.; Shu, P.; Zhang, B.; et al. Development and Validation of Machine Learning Models Based on Molecular Features for Estimating the Probability of Multiple Primary Lung Carcinoma versus Intrapulmonary Metastasis in Patients Presenting Multiple Non-Small Cell Lung Cancers. Transl. Lung Cancer Res. 2025, 14, 1118–1137. [Google Scholar] [CrossRef]
- Pang, A.W.; MacDonald, J.R.; Pinto, D.; Wei, J.; Rafiq, M.A.; Conrad, D.F.; Park, H.; Hurles, M.E.; Lee, C.; Venter, J.C.; et al. Towards a Comprehensive Structural Variation Map of an Individual Human Genome. Genome Biol. 2010, 11, R52. [Google Scholar] [CrossRef]
- Duan, G.; Huo, Q.; Ni, W.; Ding, F.; Ye, Y.; Tang, T.; Dai, H. Integrative Machine Learning Model for Subtype Identification and Prognostic Prediction in Lung Squamous Cell Carcinoma. Discov. Onc 2025, 16, 886. [Google Scholar] [CrossRef]
- Lareau, C.A.; Duarte, F.M.; Chew, J.G.; Kartha, V.K.; Burkett, Z.D.; Kohlway, A.S.; Pokholok, D.; Aryee, M.J.; Steemers, F.J.; Lebofsky, R.; et al. Droplet-Based Combinatorial Indexing for Massive-Scale Single-Cell Chromatin Accessibility. Nat. Biotechnol. 2019, 37, 916–924. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Várnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-Cycle Dynamics of Chromosomal Organization at Single-Cell Resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the Three-Dimensional Organization of Genomes: Interpreting Chromatin Interaction Data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Lv, X.; Ying, S.; Guo, Q. Artificial Intelligence-Driven Precision Medicine: Multi-Omics and Spatial Multi-Omics Approaches in Diffuse Large B-Cell Lymphoma (DLBCL). Front. Biosci. 2024, 29, 404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Boninsegna, L.; Yang, M.; Misteli, T.; Alber, F.; Ma, J. Computational Methods for Analysing Multiscale 3D Genome Organization. Nat. Rev. Genet. 2024, 25, 123–141. [Google Scholar] [CrossRef]
- Van Karnebeek, C.D.M.; Wortmann, S.B.; Tarailo-Graovac, M.; Langeveld, M.; Ferreira, C.R.; Van De Kamp, J.M.; Hollak, C.E.; Wasserman, W.W.; Waterham, H.R.; Wevers, R.A.; et al. The Role of the Clinician in the Multi-omics Era: Are You Ready? J. Inher Metab. Disea 2018, 41, 571–582. [Google Scholar] [CrossRef]
- O’Connor, O.; McVeigh, T.P. Increasing Use of Artificial Intelligence in Genomic Medicine for Cancer Care- the Promise and Potential Pitfalls. BJC Rep. 2025, 3, 20. [Google Scholar] [CrossRef]
- Lin, M.; Guo, J.; Gu, Z.; Tang, W.; Tao, H.; You, S.; Jia, D.; Sun, Y.; Jia, P. Machine Learning and Multi-Omics Integration: Advancing Cardiovascular Translational Research and Clinical Practice. J. Transl. Med. 2025, 23, 388. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Description | Types of Rearrangements | Associated Disorders |
|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | Error-prone repair of DNA double-strand breaks without homology | Translocations, deletions, insertions | Leukaemias (e.g., BCR::ABL1), lymphomas |
| Microhomology-Mediated End Joining (MMEJ) | Alternative end joining using short homologous sequences (microhomologies) | Small deletions and complex rearrangements | Genetic syndromes with structural variation |
| Fork Stalling and Template Switching (FoSTeS) | Template switching during replication fork stalling | Complex duplications and deletions | Genomic disorders (e.g., MECP2 duplication syndrome) |
| Non-Allelic Homologous Recombination (NAHR) | Recombination between low copy repeats leading to misalignment | Recurrent deletions, duplications, inversions | DiGeorge syndrome (22q11.2), Williams syndrome |
| Chromothripsis | Massive chromosome fragmentation and disordered reassembly | Complex rearrangements | Aggressive solid tumors, neuroblastoma |
| Chromoanasynthesis | Replication-based mechanism involving serial template switching and fork collapse | Complex duplications, deletions, triplications | Neurodevelopmental disorders, congenital anomalies |
| Chromoplexy | Interdependent chromosomal translocations and deletions, often in a single event | Balanced and unbalanced rearrangements | Prostate cancer, other solid tumors |
| Replication Timing (a) and Band Staining Properties (b) | |||||
|---|---|---|---|---|---|
| Features | Very Early (a) R-Positive (b) | Early (a) R-Positive (b) | Late (a) R-Negative (b) | Very Late (a) R-Negative (b) | |
| Replication category (c) | 1–3 | 4–9 | 10–14 | 15–19 | [64,72] |
| GC content | >52% | 44–52% | 37–44% | <37% | [74] |
| Gene density | >15 genes/Mb | 5–15 genes/Mb | 5–15 genes/Mb | <5 genes/Mb | [75,76] |
| General chromatin state | Highly decondensed | Variable (d) | Variable (d) | Highly condensed | [10,66] |
| General transcriptional activity | High | Variable (d) | Variable (d) | Low | [63] |
| Intranuclear localization | Internal | Intermediate | Intermediate | Peripheral | [14,77] |
| Sensitivity to replication stress | Low | Intermediate | Intermediate | High | [78] |
| Technique | Resolution | Type of Variant Detected | Main Applications | Main Limitations | Clinical Use |
|---|---|---|---|---|---|
| Classical Cytogenetics (Banding) | ~5–10 Mb | Numerical abnormalities, large unbalanced or balanced rearrangements | Chromosomal syndromes; constitutional and acquired translocations | Low resolution; subjective interpretation; limited to dividing cells | Prenatal, hematology, infertility |
| FISH | ~100 kb | Targeted deletions, duplications, translocations | Rapid targeted diagnosis; monitoring known aberrations | Not genome-wide; probe design required | Oncology, known microdeletions |
| Array-CGH | ~10–50 kb | Submicroscopic copy number variations (CNVs) | Diagnosis of genomic syndromes; oncogenomic profiling | Cannot detect balanced rearrangements | Syndromic anomalies, prenatal testing |
| SNP Arrays | ~10–50 kb | CNVs, copy-neutral LOH, uniparental disomy | Diagnosis of genetic diseases; tumor susceptibility screening | Limited for structural complexity; no balanced rearrangements | Oncology |
| Optical Mapping | kb–Mb | Complex rearrangements, large insertions, repeats | Cancer diagnostics; structural variant resolution in genetic disorders | Still emerging; requires high molecular weight DNA | Cancer, constitutional SV screening |
| Short-Read Sequencing | bp–kb | SNVs, indels, micro-CNVs | Exome/genome sequencing; detailed molecular diagnostics | Poor detection of structural variants in repetitive regions | Rare diseases, cancer genomics |
| Long-Read Sequencing | kb–Mb | Complex structural variants, repeat expansions, inversions | Rare disease diagnosis; structural variant characterization; research | High cost; error rates (platform-dependent); large data volume | Complex cases, unresolved diagnostics |
| Cytogenomic Alteration | Tumor Type | Mechanism | Clinical Implications | References |
|---|---|---|---|---|
| t(9;22)(q34;q11)—BCR::ABL1 | Chronic myeloid leukemia | Balanced translocation and fusion gene | Diagnostic marker; targeted therapy with tyrosine kinase inhibitors | [113,114] |
| t(14;18)(q32;q21)—BCL2 | Follicular lymphoma | Balanced translocation | Diagnostic and prognostic marker | [115,116] |
| Amplifications and rearrangements of MYC | Aggressive lymphomas, solid tumors | Amplifications and translocations | Poor prognosis; oncogenic activation | [117] |
| Chromothripsis | Solid and hematological malignancies | Complex genome rearrangement | Genomic instability; unfavorable prognosis | [41] |
| Enhancer hijacking | Leukaemias, solid tumors | Oncogene activation via aberrant enhancer contacts | Novel diagnostic and therapeutic targets | [7,118] |
| 5q deletions | Myelodysplastic syndrome, acute leukemia | Genomic loss | Loss of tumor suppressor genes; prognostic significance | [119] |
| inv(16)(p13q22) | Acute myeloid leukemia | Chromosomal inversion and fusion gene | Favorable prognosis; diagnostic marker | [120] |
| del(17p)—TP53 | Various tumors and leukaemias | Tumor suppressor gene loss | Poor prognosis; therapy resistance | [121] |
| Technique | Principle | Clinical Applications | Strengths | Limitations |
|---|---|---|---|---|
| Hi-C | Genome-wide sequencing of chromatin interactions | Detection of altered TADs, chromatin loops, and rearrangements in cancer and genetic diseases | Genome-wide coverage, 3D spatial information | Limited resolution without enrichment |
| Capture Hi-C | Targeted enrichment of Hi-C libraries | High-resolution analysis of disease-associated loci | Target specificity, high resolution | Dependent on prior selection of target regions |
| ChIP-seq | Sequencing of DNA bound to histone marks or TFs via immunoprecipitation | Epigenetic profiling in cancers and developmental disorders | Functional annotation of regulatory states | Requires specific antibodies and good-quality samples |
| ATAC-seq | Tagging of open chromatin with Tn5 transposase | Active regulatory region identification, tumor heterogeneity | Low input, high resolution | No protein-specific information |
| HiChIP/PLAC-seq | Combination of Hi-C and ChIP-seq to map protein-mediated chromatin contacts | Enhancer–promoter mapping in rare diseases and cancer | Protein-centric 3D contact profiling | Technically complex and expensive |
| Single-cell Hi-C/ATAC-seq | Cell-level profiling of 3D structure or chromatin accessibility | Heterogeneity analysis in cancer and rare disease | Single-cell resolution, fine population analysis | High cost, complex data analysis |
| DNA FISH | Fluorescent hybridization of DNA probes on cells or nuclei | Rearrangement detection, nuclear positioning analysis in cancers | Visual validation, widely used in diagnostics | Limited resolution, not genome wide |
| CUT&Tag/CUT&RUN | Epigenetic profiling with minimal material input | Rapid profiling in clinical specimens | Low input requirements, high sensitivity | Protocol standardization still required for clinical use |
| Optical Mapping | High-resolution physical mapping of long DNA molecules | Detection of complex rearrangements, integration with 3D genome data | Complementary to sequencing, provides structural insight | Lacks direct 3D spatial data |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Federico, C.; Brancato, D.; Bruno, F.; Coniglio, E.; Sturiale, V.; Saccone, S. Cytogenetics and Cytogenomics in Clinical Diagnostics: Genome Architecture, Structural Variants, and Translational Applications. Genes 2025, 16, 780. https://doi.org/10.3390/genes16070780
Federico C, Brancato D, Bruno F, Coniglio E, Sturiale V, Saccone S. Cytogenetics and Cytogenomics in Clinical Diagnostics: Genome Architecture, Structural Variants, and Translational Applications. Genes. 2025; 16(7):780. https://doi.org/10.3390/genes16070780
Chicago/Turabian StyleFederico, Concetta, Desiree Brancato, Francesca Bruno, Elvira Coniglio, Valentina Sturiale, and Salvatore Saccone. 2025. "Cytogenetics and Cytogenomics in Clinical Diagnostics: Genome Architecture, Structural Variants, and Translational Applications" Genes 16, no. 7: 780. https://doi.org/10.3390/genes16070780
APA StyleFederico, C., Brancato, D., Bruno, F., Coniglio, E., Sturiale, V., & Saccone, S. (2025). Cytogenetics and Cytogenomics in Clinical Diagnostics: Genome Architecture, Structural Variants, and Translational Applications. Genes, 16(7), 780. https://doi.org/10.3390/genes16070780