
Novel Pathogenic Variant c.258A>C, p.(Glu86Asp) in the TTR Gene in a Bulgarian Patient with Hereditary Transthyretin Amyloidosis

 , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
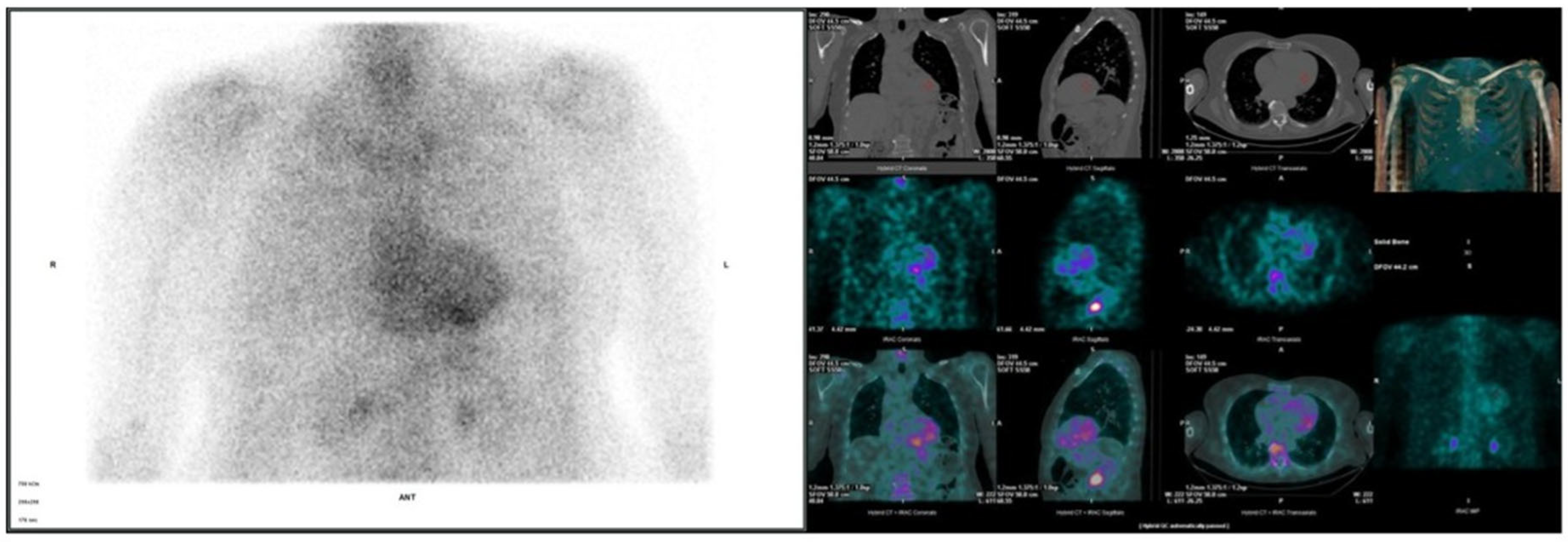
3. Results
3.1. Physical Examination
3.2. Cardiac Evaluation
3.3. Neurological Evaluation
3.4. Genetic Testing
3.5. Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef]
- Chamova, T.; Gospodinova, M.; Asenov, O.; Todorov, T.; Pavlova, Z.; Kirov, A.; Cherninkova, S.; Kastreva, K.; Taneva, A.; Blagoeva, S.; et al. Seven Years of Selective Genetic Screening Program and Follow-Up of Asymptomatic Carriers with Hereditary Transthyretin Amyloidosis in Bulgaria. Front. Neurol. 2022, 13, 844595. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-i.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef]
- Obi, C.A.; Mostertz, W.C.; Griffin, J.M.; Judge, D.P. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist. Debakey Cardiovasc. J. 2022, 18, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Fenu, S. The tip of the iceberg in ATTRv: When to start carrier monitoring and when to initiate treatment? J. Neurol. Neurosurg. Psychiatry 2024, 95, 487. [Google Scholar] [CrossRef]
- Beauvais, D.; Labeyrie, C.; Cauquil, C.; Francou, B.; Eliahou, L.; Not, A.; Echaniz-Laguna, A.; Adam, C.; Slama, M.S.; Benmalek, A.; et al. Detailed clinical, physiological and pathological phenotyping can impact access to disease-modifying treatments in ATTR carriers. J. Neurol. Neurosurg. Psychiatry 2024, 95, 489–499. [Google Scholar] [CrossRef]
- Brito, D.; Albrecht, F.C.; Arenaza, D.P.; Bart, N.; Better, N.; Carvajal-Juarez, I.; Conceição, I.; Damy, T.; Dorbala, S.; Fidalgo, J.-C.; et al. World Heart Federation Consensus on Transthyretin Amyloidosis Cardiomyopathy (ATTR-CM). Glob. Heart 2023, 18, 59. [Google Scholar] [CrossRef]
- Rozenbaum, M.H.; Large, S.; Bhambri, R.; Stewart, M.; Whelan, J.; van Doornewaard, A.; Dasgupta, N.; Masri, A.; Nativi-Nicolau, J. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol. Ther. 2021, 10, 141–159. [Google Scholar] [CrossRef]
- Rosenblum, H.; Castano, A.; Alvarez, J.; Goldsmith, J.; Helmke, S.; Maurer, M.S. TTR (Transthyretin) Stabilizers Are Associated with Improved Survival in Patients with TTR Cardiac Amyloidosis. Circ. Hear. Fail. 2018, 11, e004769. [Google Scholar] [CrossRef]
- Keller, D.M.; Straburzyńska-Migaj, E.; Lesiak, M. Advancing treatments for transthyretin amyloid cardiomyopathy: Innovations in RNA silencing, gene editing, TTR stabilization, and degradation. Pol. Heart J. (Kardiol. Pol.) 2025, 83, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, Z.; Sarafov, S.; Todorov, T.; Kirov, A.; Chamova, T.; Gospodinova, M.; Tournev, I.; Mitev, V.; Todorova, A. Characterization of population genetic structure of hereditary transthyretin amyloidosis in Bulgaria. Amyloid 2021, 28, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Gospodinova, M.; Sarafov, S.; Chamova, T.; Kirov, A.; Todorov, T.; Nakov, R.; Todorova, A.; Denchev, S.; Tournev, I. Cardiac involvement, morbidity and mortality in hereditary transthyretin amyloidosis because of p.Glu89Gln mutation. J. Cardiovasc. Med. 2020, 21, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Comenzo, R.; Falk, R.H.; Fermand, J.P.; Hazenberg, B.P.; Hawkins, P.N.; Merlini, G.; Moreau, P.; Ronco, P.; Sanchorawala, V.; et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am. J. Hematol. 2005, 79, 319–328. [Google Scholar] [CrossRef]
- Dyck, P.J.; Litchy, W.J.; Lehman, K.A.; Hokanson, J.L.; Low, P.A.; O’BRien, P.C. Variables influencing neuropathic endpoints. Neurology 1995, 45, 1115–1121. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wilczek, H.E.; Nowak, G.; Larsson, M.; Oksanen, A.; Iwata, T.; Gjertsen, H.; Söderdahl, G.; Wikström, L.; Ando, Y.; et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): A single-center experience over 16 years. Am. J. Transplant. 2007, 7, 2597–2604. [Google Scholar] [CrossRef]
- Zouari, H.G.; Ng Wing Tin, S.; Wahab, A.; Damy, T.; Lefaucheur, J. Assessment of autonomic innervation of the foot in familial amyloid polyneuropathy. Eur. J. Neurol. 2019, 26, 94-e10. [Google Scholar] [CrossRef]
- Spitzer, A.; Lang, E.; Birklein, F.; Claus, D.; Neundörfer, B. Cardiac autonomic involvement and peripheral nerve function in patients with diabetic neuropathy. Funct. Neurol. 1997, 12, 115–122. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405. [Google Scholar] [CrossRef]
- Chen, Z.; Koh, J.S.; Saini, M.; Tay, K.S.S.; Jayne Tan, Y.; Chai, J.Y.H.; Fam, S.R.; Juraidah, A.; Lim, P.K.; Ng, A.S.L.; et al. Hereditary Transthyretin Amyloidosis-Clinical and Genetic Characteristics of a Multiracial South-East Asian Cohort in Singapore. J. Neuromuscul. Dis. 2021, 8, 723–733. [Google Scholar] [CrossRef]
- Lobato, L.; Rocha, A. Transthyretin amyloidosis and the kidney. Clin. J. Am. Soc. Nephrol. 2012, 7, 1337–1346. [Google Scholar] [CrossRef]
- Solignac, J.; Delmont, E.; Fortanier, E.; Attarian, S.; Mancini, J.; Daniel, L.; Ion, I.; Ricci, J.-E.; Robert, T.; Jourde-Chiche, N.; et al. Kidney involvement in hereditary transthyretin amyloidosis: A cohort study of 103 patients. Clin. Kidney J. 2022, 15, 1747–1754. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlova, Z.; Zhelyazkova, S.; Gospodinova, M.; Ormandjieva, A.; Todorov, T.; Asenov, O.; Chamova, T.; Antimov, P.; Mikova, D.; Palashev, Y.; et al. Novel Pathogenic Variant c.258A>C, p.(Glu86Asp) in the TTR Gene in a Bulgarian Patient with Hereditary Transthyretin Amyloidosis. Genes 2025, 16, 726. https://doi.org/10.3390/genes16070726
Pavlova Z, Zhelyazkova S, Gospodinova M, Ormandjieva A, Todorov T, Asenov O, Chamova T, Antimov P, Mikova D, Palashev Y, et al. Novel Pathogenic Variant c.258A>C, p.(Glu86Asp) in the TTR Gene in a Bulgarian Patient with Hereditary Transthyretin Amyloidosis. Genes. 2025; 16(7):726. https://doi.org/10.3390/genes16070726
Chicago/Turabian StylePavlova, Zornitsa, Sashka Zhelyazkova, Mariana Gospodinova, Anastasia Ormandjieva, Tihomir Todorov, Ognian Asenov, Teodora Chamova, Plamen Antimov, Dilyana Mikova, Yordan Palashev, and et al. 2025. "Novel Pathogenic Variant c.258A>C, p.(Glu86Asp) in the TTR Gene in a Bulgarian Patient with Hereditary Transthyretin Amyloidosis" Genes 16, no. 7: 726. https://doi.org/10.3390/genes16070726
APA StylePavlova, Z., Zhelyazkova, S., Gospodinova, M., Ormandjieva, A., Todorov, T., Asenov, O., Chamova, T., Antimov, P., Mikova, D., Palashev, Y., Tournev, I., & Todorova, A. (2025). Novel Pathogenic Variant c.258A>C, p.(Glu86Asp) in the TTR Gene in a Bulgarian Patient with Hereditary Transthyretin Amyloidosis. Genes, 16(7), 726. https://doi.org/10.3390/genes16070726