
A Comprehensive Review of Deep Learning Applications with Multi-Omics Data in Cancer Research

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
- Cancer type, subtype, and unknown primary origin classification;
- Driver gene prediction and discovery;
- Survival analysis;
- Drug response prediction.
2. Basic Concepts and Principles in Machine Learning and Deep Learning
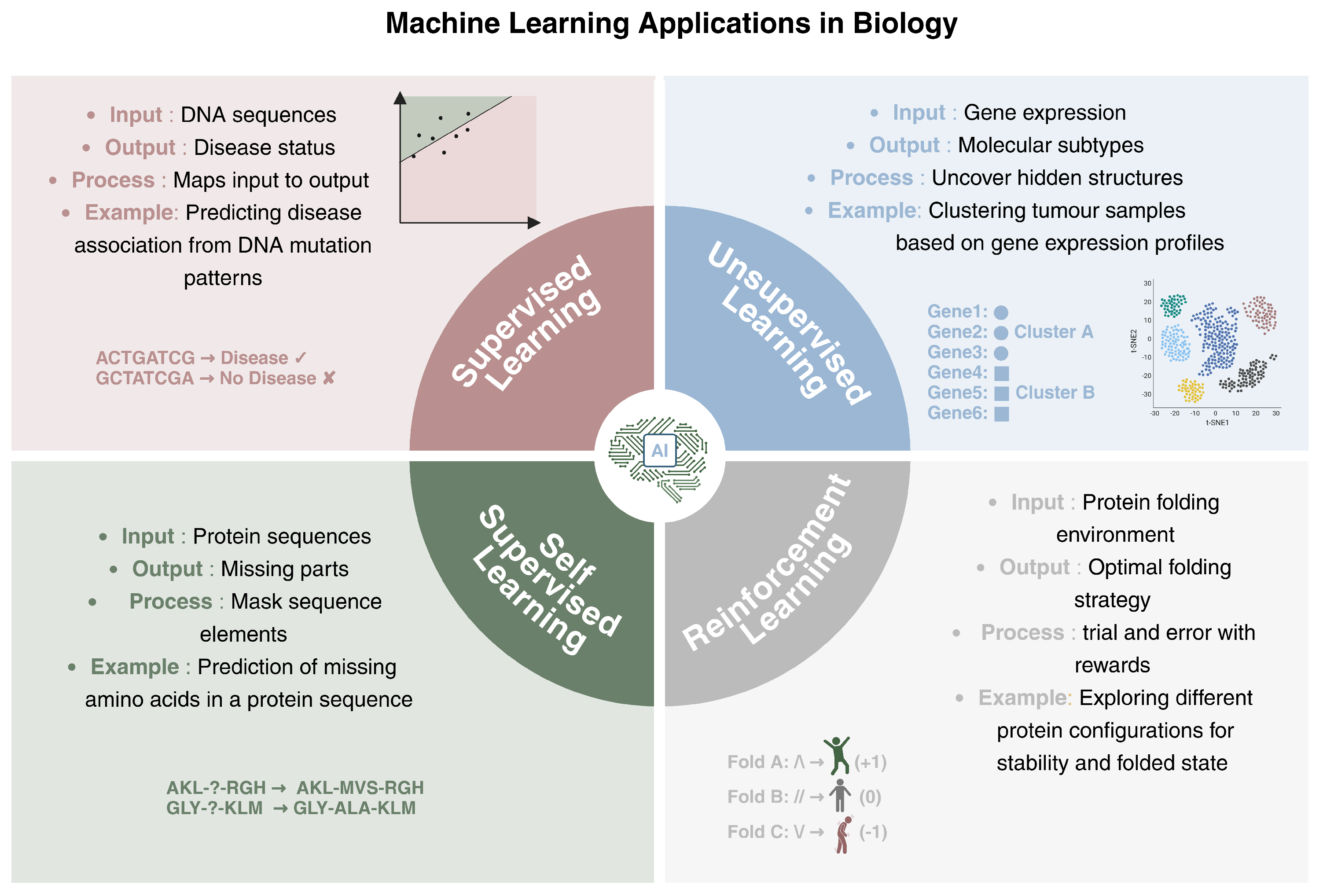
2.1. Types of Learning
- Supervised Learning: supervised models are trained on labeled data, which means that each data point is associated with a known output. The model learns how to map the inputs to their corresponding outputs, enabling it to generalize the acquired knowledge to new data. A biological example of supervised learning is predicting whether a DNA sequence with a particular mutational pattern is associated with a specific disease. The model is trained on sequences labeled as disease-associated or not, and it will learn the characteristics that differentiate the two groups.
- Unsupervised Learning: This deals with unlabeled data, so the main tasks are related to uncovering hidden structures within the data. The algorithm, through the maximization of similarity measures, identifies intrinsic patterns, without any supervision. In genomics, unsupervised learning can be applied to tasks such as clustering tumor samples based on their gene expression profiles, thus allowing for the identification of molecular subtypes. Unsupervised methods can group genes with similar expression patterns under different conditions, which may reveal insights into underlying biological processes or disease mechanisms.
- Self-Supervised Learning: This is a form of learning where the model generates its own labels from the data, rather than relying on provided labels. It often involves predicting parts of the input from other parts of the same input. This type of learning is useful when there is a large amounts of unlabeled data available. In biology, a self-supervised learning approach could involve predicting missing parts of a genomic sequence based on the context, helping the model learn useful representations of the data without requiring explicit annotations.
- Reinforcement Learning: This focuses on training agents to make a sequence of decisions by interacting with an environment. The model learns through trial and error, receiving rewards or penalties based on the actions taken. In a biological context, reinforcement learning can be applied to protein folding. A reinforcement learning model could explore different configurations of a protein sequence, receiving rewards when it folds into a more stable or functional conformation. This process mirrors how biological systems optimize complex tasks through iterations, such as how cells might improve the efficiency of molecular interactions or enzyme catalysis over time based on feedback from the system’s state.
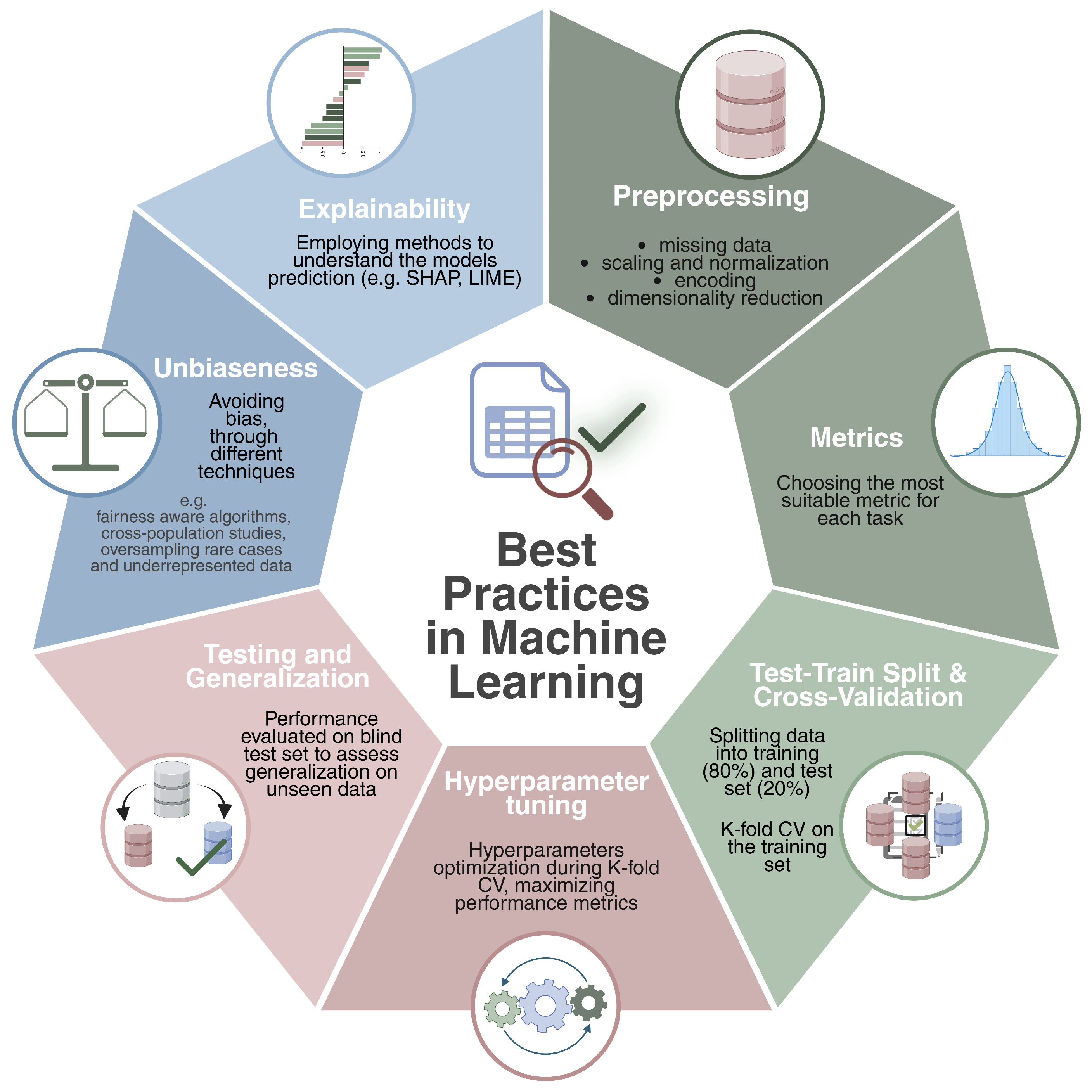
2.2. Best Practices
- Preprocessing: This is a crucial step that ensures data quality and consistency before model training. It typically involves data cleaning—handling missing values through imputation or filtering—and features preprocessing techniques such as normalization, standardization, and encoding categorical variables. Dimensionality reduction methods like PCA [43], t-SNE [44], and autoencoders [45] are often applied to mitigate the curse of dimensionality and to enhance model performance. In the context of genomic data, common practices include initial quality control—such as assessing sample integrity, read quality, and detection rates—to remove low-quality samples or features. This is followed by correcting for batch effects [46,47,48], filtering low-expressed genes in RNA-seq data, and normalizing expression levels. In DNA methylation analysis, data are often preprocessed through background correction, normalization, and filtering of low-quality probes [49,50]. For multi-omics integration, preprocessing involves aligning heterogeneous data types—such as transcriptomics, epigenomics, and proteomics—through techniques like feature harmonization and scaling, ensuring that combined signals reflect meaningful biological relationships while reducing any source of noise or bias.
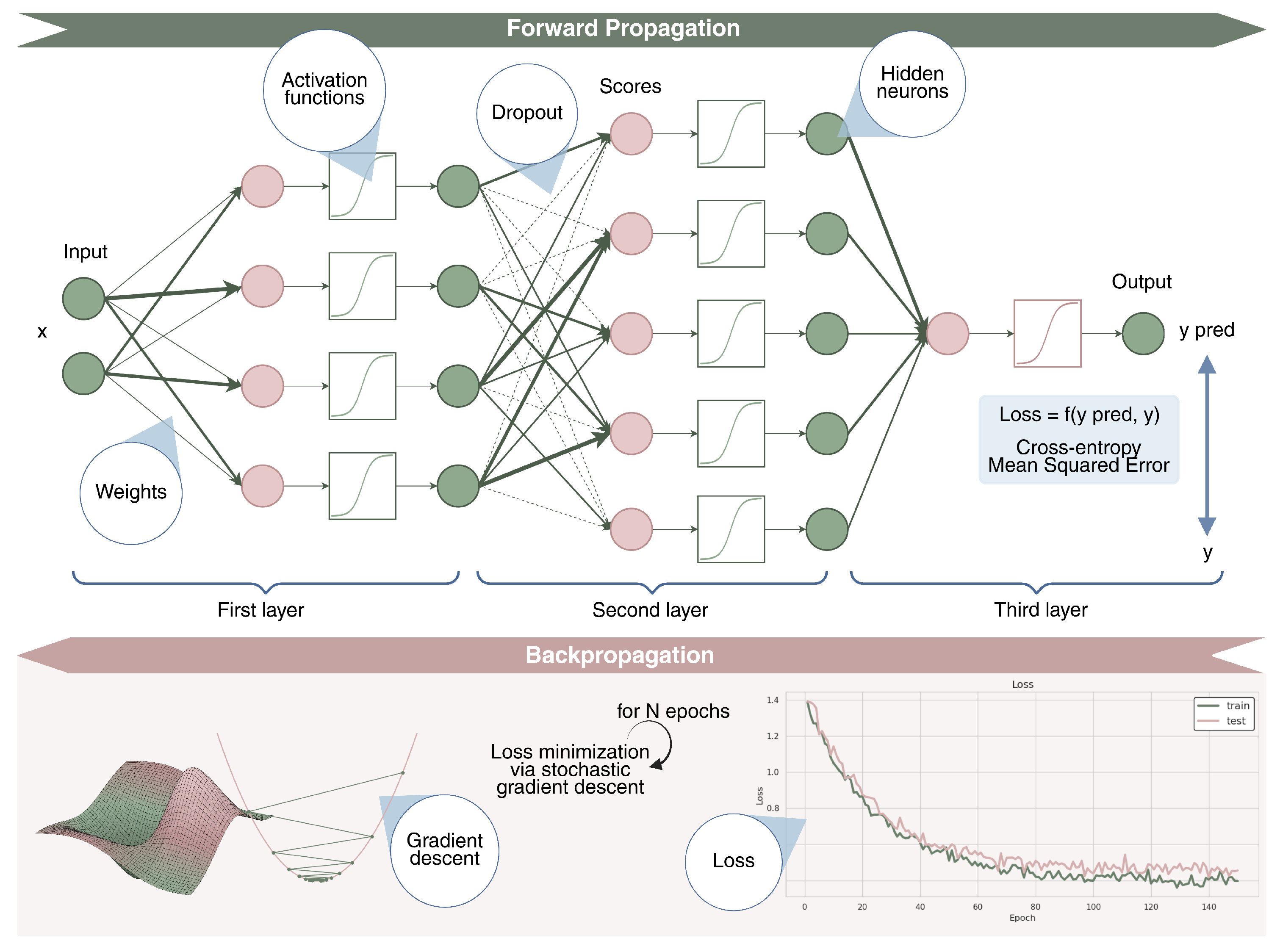
- Metrics: based on the preliminary preprocessing analysis, the nature of the problem, and the dataset, choosing the most suitable metric significantly changes the reliability and effectiveness of the implemented machine learning method. For classification tasks, common metrics include area under the curve (AUC), precision–recall area under the curve (PRAUC), sensitivity, specificity, F1-score, and Matthews correlation coefficient (MCC). To properly measure performance, it is usually good practice to provide a complete overview of the metrics, as some of them can be better suited (e.g., AUC, accuracy) to unbalanced datasets. For a complete overview and definitions of classification metrics for both binary and multi-class classification scenarios we redirect the reader to [51,52].For regression tasks, common evaluation metrics include Mean Squared Error (MSE), Mean Absolute Error (MAE), Pearson correlation (r), and the coefficient of determination (). These metrics assess the accuracy and variance explained by the model. Please refer to [53] for mathematical formulations of the common regression metrics.For survival analysis, key evaluation metrics include the Concordance Index (C-index), which quantifies the agreement between predicted and actual survival outcomes; the time-dependent Area Under the Curve (AUC), which assesses model discrimination at specific time points; and the Integrated Brier Score (IBS), which evaluates overall prediction accuracy across the entire follow-up period. A complete review of survival metrics can be found in [54]
- Train–Test Split and Cross-Validation: a common practice is to split the available dataset into training and test sets (e.g., 80/20). The training set will be used to effectively train the model and to optimize it, while the test set will serve to evaluate the model’s ability to accomplish the given task on points never seen during the training time. In particular, the training set is further divided using the k-fold cross-validation strategy. This technique estimates the model’s generalization error by dividing the training set into K subsets. folds are used for training, while the remaining fold is used to validate the prediction by calculating a specific evaluation metric. This process is repeated K times, ensuring that each fold is used as a validation set once. The average validation error across all iterations provides a robust estimate of model performance. Cross-validation is commonly employed for hyperparameter tuning, but in the absence of an external blind test set, it can also serve as a surrogate for model evaluation, providing an estimate of generalization performance.However, depending on the nature of the problem and the specific task, it might be beneficial to use stratified K-fold cross-validation instead of a standard K-fold approach. This is particularly useful when dealing with imbalanced datasets, where certain classes are underrepresented. In a traditional K-fold split, the distribution of classes in each fold may not be representative of the overall dataset, potentially leading to biased training and evaluation. Stratified K-fold addresses this issue by preserving the class distribution within each fold, ensuring that each subset maintains a proportionally similar representation of the different categories present in the dataset. This is crucial in classification tasks, as it prevents the model from overfitting to the majority class while underperforming on minority classes.
- Hyperparameter Tuning: each ML algorithm (e.g., random forest, support vector machines) has specific hyperparameters (e.g., number of trees, penalty) that should be optimized to obtain the best-performing model. During the cross-validation, a range of hyperparameter values is tested to determine the configuration that optimizes the user’s predefined metrics. This ensures that the selected model is the optimal one for the given task based on the desired metrics.
- Testing and Generalization: once the optimal model is identified, its final evaluation is performed on the test set, which was not used during training. This step measures the model’s ability to generalize to new data, ensuring it can obtain accurate predictions in real-world data.
- Unbiasness: bias and fairness remain critical challenges in machine learning [55], often arising from different sources such as imbalanced datasets, measurement inconsistencies and annotation errors. In the field of biology, for example, limited genetic diversity in reference databases or the under-representation of rare diseases can lead to biased models that fail to provide accurate predictions for minority groups. Fairness-aware algorithms, such as adversarial debiasing [56], tackle these issues by training models to minimize reliance on sensitive attributes, such as demographic factors, while maintaining high prediction accuracy. Additionally, cross-population validation studies enable models to be evaluated on diverse and independent population groups, ensuring that predictions generalize effectively. By combining these approaches with techniques like oversampling rare cases [57] and reweighting underrepresented data, it becomes possible to achieve robust and reliable performance in biological applications.
- Explainability: understanding the challenges of explainability in machine learning is essential, particularly in the context of biology and clinical applications. One significant challenge is the difficulty of explaining how models capture biological mechanisms and how predictions can be interpreted, especially for large datasets. Additionally, the transparency of highly accurate methods, such as deep learning, is often limited, while interpretable models like decision trees may lack the performance needed for complex tasks [58]. These limitations come from the way our brains work, making it hard for us to fully understand how models make decisions or what factors influence them [59]. Despite these challenges, significant advances have been made in developing tools like SHAP [60] and LIME [61]. These model-agnostic tools improve transparency by providing approximations of how models make decisions. They work by explaining the model’s behavior and identifying the factors that influence its predictions. Through techniques like visualizations and feature importance scores, these tools help users to understand which inputs are most impactful, making it easier to trust and understand the model’s decisions and ensuring safety in critical applications. In tasks involving image data—such as histopathology or medical imaging—techniques as saliency maps [62], Grad-CAM [63], and attention weight visualization have become valuable. These methods highlight the regions in an image that most contribute to the model decision, offering intuitive insights into model behavior. Through such visualizations and feature importance scores, these tools help users understand which inputs are most impactful.
2.3. Deep Learning
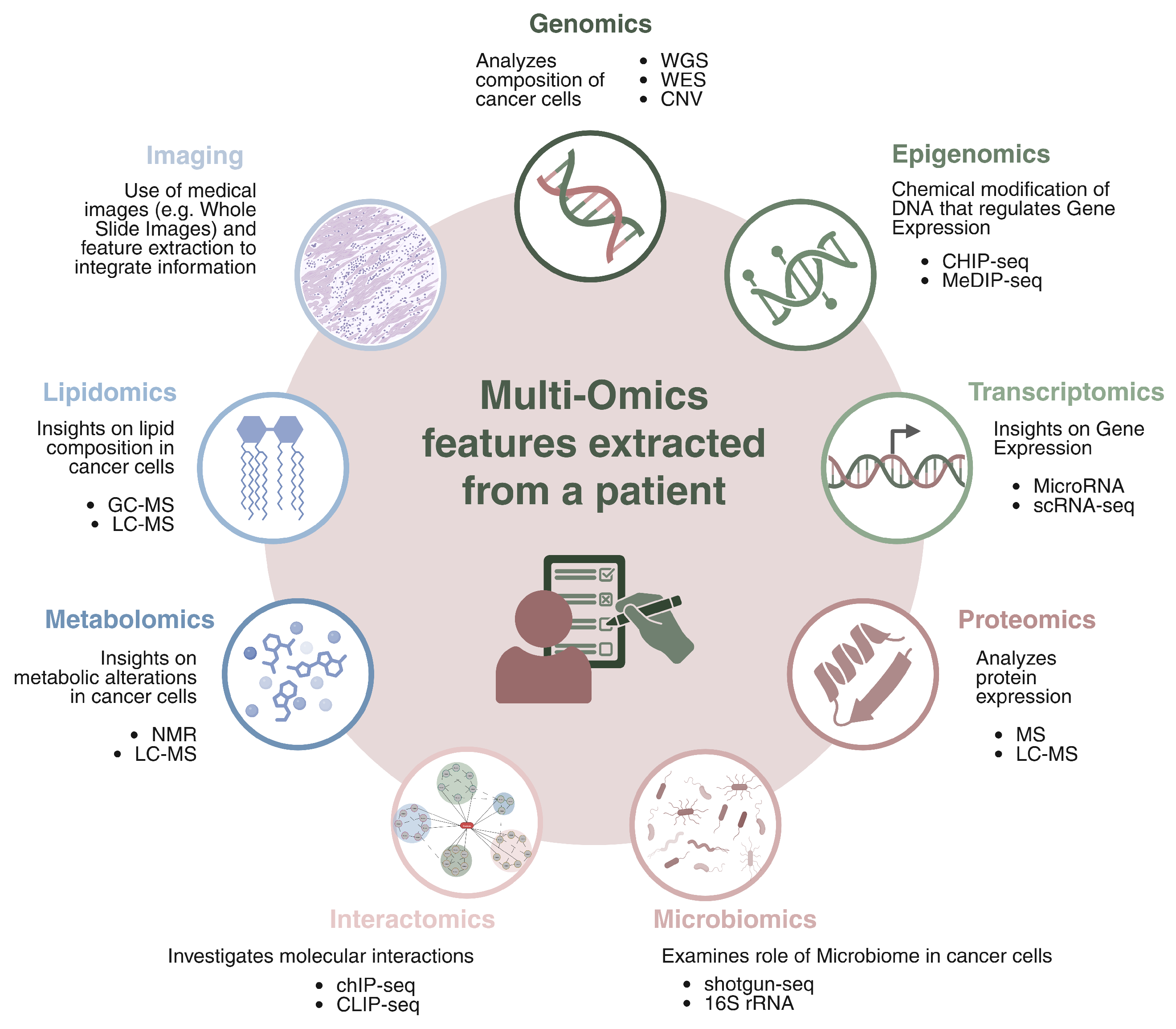
3. Introduction to Multi-Omics
- Genomics focuses on the comprehensive analysis of the genetic landscape of cancer cells, with the goal of identifying mutations, structural variations, and alterations that drive tumor initiation and progression. Technologies such as Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES) are fundamental for profiling both coding and non-coding regions of the genome. These approaches enable the detection of somatic alterations including Single and Multiple Nucleotide Variants (SNVs, MNVs), insertions and deletions (indels), Copy Number Variations (CNVs), and more complex structural events. Additionally, genomic data are used to assess Microsatellite Instability (MSI), which reflects defects in the DNA mismatch repair system, and to study others genomic properties that can help to better profile cancer.
- Epigenomics investigates chemical modifications of DNA and histones that regulate gene expression without altering the underlying DNA sequence. Epigenetic changes, such as DNA methylation, histone modifications, and chromatin remodeling, are responsible of controlling gene accessibility and transcriptional activity. These modifications affect gene silencing, activation, and cellular differentiation, and they are often dysregulated in cancer. Aberrant epigenetic patterns can lead to the inactivation of tumor suppressor genes or the activation of oncogenes, contributing to cancer initiation and progression. Key epigenomic techniques include DNA methylation analysis (e.g., MeDIP-seq [69] and Bisulfite sequencing [70]), histone modification profiling (e.g., ChIP-seq [71]), and chromatin accessibility assays (e.g., ATAC-seq [72]), all of which help to understand how chromatin structure and gene expression are altered in cancer.
- Transcriptomics provides insight into gene expression patterns by analyzing RNA molecules, offering a snapshot of how genetic information is transcribed and regulated. Key methods in transcriptomics include RNA Sequencing (RNA-seq [73]) for global gene expression profiling, Long Non-Coding RNA (lncRNA-seq [74]) for the study of non-coding RNAs involved in gene regulation, MicroRNA (miRNA-seq [75]) to identify small RNAs regulating mRNA stability and translation, and single-sell RNA-seq (scRNA-seq [76]) for profiling gene expression. Additionally, spatial transcriptomics allows for the mapping of RNA expression within tissue architecture, providing insights into the tumor microenvironment.
- Proteomics is the comprehensive study of the full set of proteins produced in a cell, tissue, or organism, focusing on their expression levels, structural modifications, and interactions. It provides valuable insight into cancer research by providing information on how protein dynamics change during cancer development and progression. One of the key technologies in proteomics is mass spectrometry, including methods like tandem mass spectrometry (MS/MS [77]) and liquid chromatography–mass spectrometry (LC-MS [78]), which enable precise identification and quantification of proteins in samples. Proteomics also allows for the mapping of protein–protein interaction networks, which are essential for understanding the molecular pathways involved in cancer. A critical aspect of this field is the investigation of post-translational modifications such as phosphorylation, ubiquitination, and acetylation, which influence protein function and are often dysregulated in cancer.
- Metabolomics studies the metabolic alterations in cancer cells, analyzing small molecules involved in cellular metabolism. Techniques such as Nuclear Magnetic Resonance (NMR [79]) spectroscopy and LC-MS are used for the profiling and quantification of intracellular and extracellular metabolites, providing insights into tumor metabolism, progression, and drug response.
- Lipidomics explores the lipid composition of cancer cells, examining the role of lipids in cancer metabolism and progression. By characterizing lipid profiles through techniques like LC-MS technology, Gas Chromatography–Mass Spectrometry (GC-MS) [80], and shotgun lipidomics [81], it is possible to uncover the significance of lipid metabolism in tumor biology.
- Microbiomics examines the role of the microbiome in cancer development and therapy response, highlighting the importance of microbial communities in the tumor microenvironment. 16S rRNA sequencing and metagenomics (shotgun sequencing) are used to profile the microbiota associated with tumors, providing insights into how the microbiome may influence cancer progression and therapeutic outcomes.
- Interactomics investigates molecular interactions, such as protein–DNA, protein–RNA, and protein–protein interactions, to map the regulatory networks driving cancer. Techniques like ChIP-seq [82] and CUT&RUN [83] are used to analyze protein–DNA interactions, while CLIP-seq [84] and RIP-seq [85] explore protein–RNA interactions. Protein–protein interactions, mapped through methods like Yeast Two-Hybrid (Y2H) [86] reveal the complex networks of molecular interactions driving cancer biology.
- Imaging omics merges medical imaging and molecular data, extracting quantitative features from imaging modalities (e.g., H&E, CT, MRI, PET) and integrating them with molecular data for a more comprehensive understanding of tumor behavior. The use of AI to analyze digital pathology slides is at the forefront of this field, enabling more accurate cancer diagnosis and prognosis.
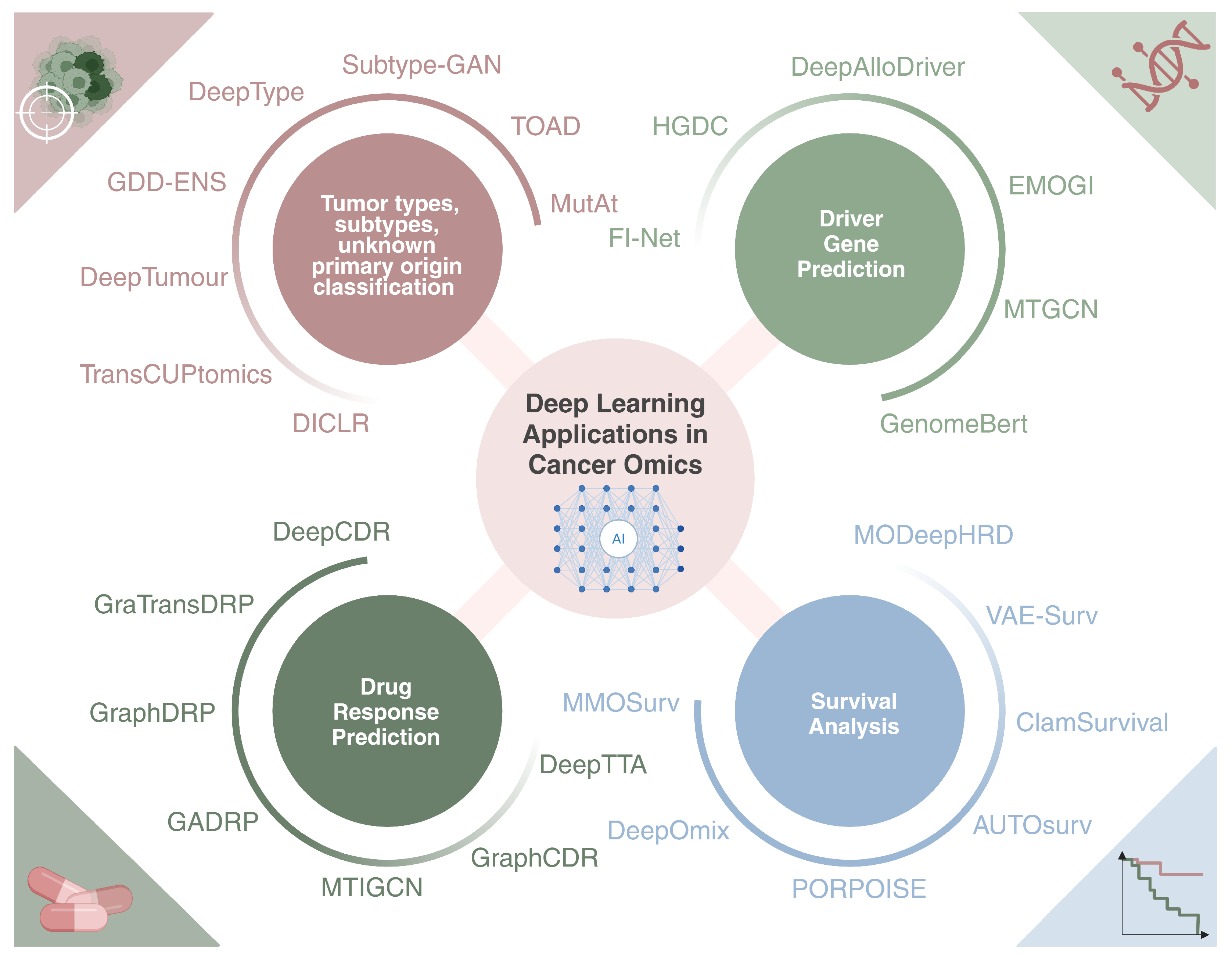
4. Deep Learning Applications in Cancer Omics
4.1. Classification of Tumor Types, Subtypes, and Unknown Primary Origin
4.2. Driver Gene Prediction
4.3. Multi-Omics Survival Analysis
4.4. Drug Response Prediction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Quazi, S. Artificial intelligence and machine learning in precision and genomic medicine. Med. Oncol. 2022, 39, 120. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Pancotti, C.; Rollo, C.; Codicè, F.; Birolo, G.; Fariselli, P.; Sanavia, T. MUSE-XAE: MUtational Signature Extraction with eXplainable AutoEncoder enhances tumour types classification. Bioinformatics 2024, 40, btae320. [Google Scholar] [CrossRef]
- Manders, F.; Brandsma, A.M.; de Kanter, J.; Verheul, M.; Oka, R.; van Roosmalen, M.J.; van der Roest, B.; van Hoeck, A.; Cuppen, E.; van Boxtel, R. MutationalPatterns: The one stop shop for the analysis of mutational processes. BMC Genom. 2022, 23, 134. [Google Scholar] [CrossRef]
- Islam, S.A.; Díaz-Gay, M.; Wu, Y.; Barnes, M.; Vangara, R.; Bergstrom, E.N.; He, Y.; Vella, M.; Wang, J.; Teague, J.W.; et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2022, 2, 100179. [Google Scholar] [CrossRef]
- Pancotti, C.; Rollo, C.; Birolo, G.; Benevenuta, S.; Fariselli, P.; Sanavia, T. Unravelling the instability of mutational signatures extraction via archetypal analysis. Front. Genet. 2023, 13, 1049501. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant olaparib for patients with BRCA1-or BRCA2-mutated breast cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Levatić, J.; Salvadores, M.; Fuster-Tormo, F.; Supek, F. Mutational signatures are markers of drug sensitivity of cancer cells. Nat. Commun. 2022, 13, 2926. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K. An introduction to convolutional neural networks. arXiv 2015, arXiv:1511.08458. [Google Scholar]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Medsker, L.R.; Jain, L. Recurrent neural networks. Des. Appl. 2001, 5, 2. [Google Scholar]
- Vaswani, A. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 6000–6010. [Google Scholar]
- Tasaki, S.; Gaiteri, C.; Mostafavi, S.; Wang, Y. Deep learning decodes the principles of differential gene expression. Nat. Mach. Intell. 2020, 2, 376–386. [Google Scholar] [CrossRef]
- Sekhon, A.; Singh, R.; Qi, Y. DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications. Bioinformatics 2018, 34, i891–i900. [Google Scholar] [CrossRef]
- Sun, P.; Fan, S.; Li, S.; Zhao, Y.; Lu, C.; Wong, K.C.; Li, X. Automated exploitation of deep learning for cancer patient stratification across multiple types. Bioinformatics 2023, 39, btad654. [Google Scholar] [CrossRef]
- Zhou, J.; Deirawan, H.; Daaboul, F.; Aung, T.N.; Beydoun, R.; Ahmed, F.S.; Chuang, J.H.; Foroughi, A. Integrative deep learning analysis improves colon adenocarcinoma patient stratification at risk for mortality. EBioMedicine 2023, 94, 104726. [Google Scholar] [CrossRef]
- Azuaje, F. Artificial intelligence for precision oncology: Beyond patient stratification. NPJ Precis. Oncol. 2019, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Cheerla, A.; Gevaert, O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics 2019, 35, i446–i454. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhan, X.; Xiang, S.; Johnson, T.S.; Helm, B.; Yu, C.Y.; Zhang, J.; Salama, P.; Rizkalla, M.; Han, Z.; et al. SALMON: Survival analysis learning with multi-omics neural networks on breast cancer. Front. Genet. 2019, 10, 166. [Google Scholar] [CrossRef]
- D’Amico, S.; Dall’Olio, L.; Rollo, C.; Alonso, P.; Prada-Luengo, I.; Dall’Olio, D.; Sala, C.; Sauta, E.; Asti, G.; Lanino, L.; et al. MOSAIC: An Artificial Intelligence–Based Framework for Multimodal Analysis, Classification, and Personalized Prognostic Assessment in Rare Cancers. JCO Clin. Cancer Inform. 2024, 8, e2400008. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Krishnamachari, K.; Lu, D.; Swift-Scott, A.; Yeraliyev, A.; Lee, K.; Huang, W.; Leng, S.N.; Skanderup, A.J. Accurate somatic variant detection using weakly supervised deep learning. Nat. Commun. 2022, 13, 4248. [Google Scholar] [CrossRef]
- Yassi, M.; Chatterjee, A.; Parry, M. Application of deep learning in cancer epigenetics through DNA methylation analysis. Brief. Bioinform. 2023, 24, bbad411. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Sharma, H.; Zerbe, N.; Klempert, I.; Hellwich, O.; Hufnagl, P. Deep convolutional neural networks for automatic classification of gastric carcinoma using whole slide images in digital histopathology. Comput. Med. Imaging Graph. 2017, 61, 2–13. [Google Scholar] [CrossRef]
- Xu, H.; Usuyama, N.; Bagga, J.; Zhang, S.; Rao, R.; Naumann, T.; Wong, C.; Gero, Z.; González, J.; Gu, Y.; et al. A whole-slide foundation model for digital pathology from real-world data. Nature 2024, 630, 181–188. [Google Scholar] [CrossRef]
- Lu, M.Y.; Chen, T.Y.; Williamson, D.F.; Zhao, M.; Shady, M.; Lipkova, J.; Mahmood, F. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021, 594, 106–110. [Google Scholar] [CrossRef]
- Huang, K.; Lin, B.; Liu, J.; Liu, Y.; Li, J.; Tian, G.; Yang, J. Predicting colorectal cancer tumor mutational burden from histopathological images and clinical information using multi-modal deep learning. Bioinformatics 2022, 38, 5108–5115. [Google Scholar] [CrossRef]
- Mondol, R.K.; Millar, E.K.; Graham, P.H.; Browne, L.; Sowmya, A.; Meijering, E. hist2rna: An efficient deep learning architecture to predict gene expression from breast cancer histopathology images. Cancers 2023, 15, 2569. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Abbasi, A.; Díaz-Gay, M.; Galland, L.; Ladoire, S.; Lippman, S.M.; Alexandrov, L.B. Deep learning artificial intelligence predicts homologous recombination deficiency and platinum response from histologic slides. J. Clin. Oncol. 2024, 42, 3550–3560. [Google Scholar] [CrossRef]
- John, A.; Qin, B.; Kalari, K.R.; Wang, L.; Yu, J. Patient-specific multi-omics models and the application in personalized combination therapy. Future Oncol. 2020, 16, 1737–1750. [Google Scholar] [CrossRef]
- Koh, L.; Novera, W.; Lim, S.W.; Chong, Y.K.; Pang, Q.Y.; Low, D.; Ang, B.T.; Tang, C. Integrative multi-omics approach to targeted therapy for glioblastoma. Pharmacol. Res. 2022, 182, 106308. [Google Scholar] [CrossRef]
- Chakraborty, S.; Sharma, G.; Karmakar, S.; Banerjee, S. Multi-OMICS approaches in cancer biology: New era in cancer therapy. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2024, 1870, 167120. [Google Scholar] [CrossRef]
- Catanese, A.; Rajkumar, S.; Sommer, D.; Masrori, P.; Hersmus, N.; Van Damme, P.; Witzel, S.; Ludolph, A.; Ho, R.; Boeckers, T.M.; et al. Multiomics and machine-learning identify novel transcriptional and mutational signatures in amyotrophic lateral sclerosis. Brain 2023, 146, 3770–3782. [Google Scholar] [CrossRef]
- O’Connor, L.M.; O’Connor, B.A.; Lim, S.B.; Zeng, J.; Lo, C.H. Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective. J. Pharm. Anal. 2023, 13, 836–850. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; van Kralingen, J.C.; Langton, K.; Lang, K.; Erlic, Z.; Larsen, C.K.; Amar, L.; Pamporaki, C.; Mulatero, P.; et al. Machine learning for classification of hypertension subtypes using multi-omics: A multi-centre, retrospective, data-driven study. EBioMedicine 2022, 84, 104276. [Google Scholar] [CrossRef]
- Doran, S.; Arif, M.; Lam, S.; Bayraktar, A.; Turkez, H.; Uhlen, M.; Boren, J.; Mardinoglu, A. Multi-omics approaches for revealing the complexity of cardiovascular disease. Brief. Bioinform. 2021, 22, bbab061. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Bank, D.; Koenigstein, N.; Giryes, R. Autoencoders. In Machine Learning for Data Science Handbook: Data Mining and Knowledge Discovery Handbook; Springer Nature: London, UK, 2023; pp. 353–374. [Google Scholar]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 1–32. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.r.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Pidsley, R.; Y Wong, C.C.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef]
- Welsh, H.; Batalha, C.; Li, W.; Mpye, K.; Souza-Pinto, N.C.d.; Naslavsky, M.; Parra, E. A systematic evaluation of normalization methods and probe replicability using infinium EPIC methylation data. Clin. Epigenet. 2023, 15, 41. [Google Scholar] [CrossRef]
- Grandini, M.; Bagli, E.; Visani, G. Metrics for multi-class classification: An overview. arXiv 2020, arXiv:2008.05756. [Google Scholar]
- Hossin, M.; Sulaiman, M.N. A review on evaluation metrics for data classification evaluations. Int. J. Data Min. Knowl. Manag. Process 2015, 5, 1. [Google Scholar]
- Naidu, G.; Zuva, T.; Sibanda, E.M. A review of evaluation metrics in machine learning algorithms. In Proceedings of the Computer Science On-Line Conference, On-Line, 3–5 April 2023; pp. 15–25. [Google Scholar]
- Suresh, K.; Severn, C.; Ghosh, D. Survival prediction models: An introduction to discrete-time modeling. BMC Med. Res. Methodol. 2022, 22, 207. [Google Scholar] [CrossRef]
- Mehrabi, N.; Morstatter, F.; Saxena, N.; Lerman, K.; Galstyan, A. A survey on bias and fairness in machine learning. ACM Comput. Surv. (CSUR) 2021, 54, 1–35. [Google Scholar] [CrossRef]
- Zhang, B.H.; Lemoine, B.; Mitchell, M. Mitigating unwanted biases with adversarial learning. In Proceedings of the 2018 AAAI/ACM Conference on AI, Ethics, and Society, New Orleans, LA, USA, 2–3 February 2018; pp. 335–340. [Google Scholar]
- Mohammed, R.; Rawashdeh, J.; Abdullah, M. Machine learning with oversampling and undersampling techniques: Overview study and experimental results. In Proceedings of the 2020 11th International Conference on Information and Communication Systems (ICICS), Irbid, Jordan, 7–9 April 2020; pp. 243–248. [Google Scholar]
- Bologna, G.; Hayashi, Y. Characterization of symbolic rules embedded in deep DIMLP networks: A challenge to transparency of deep learning. J. Artif. Intell. Soft Comput. Res. 2017, 7, 265–286. [Google Scholar] [CrossRef]
- Peters, J.; Janzing, D.; Schölkopf, B. Elements of Causal Inference: Foundations and Learning Algorithms; The MIT Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Lundberg, S. A unified approach to interpreting model predictions. arXiv 2017, arXiv:1705.07874. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why should i trust you?” Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep inside convolutional networks: Visualising image classification models and saliency maps. arXiv 2013, arXiv:1312.6034. [Google Scholar]
- Selvaraju, R.R.; Das, A.; Vedantam, R.; Cogswell, M.; Parikh, D.; Batra, D. Grad-CAM: Why did you say that? arXiv 2016, arXiv:1611.07450. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Amari, S.i. Backpropagation and stochastic gradient descent method. Neurocomputing 1993, 5, 185–196. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Hinton, G.; Srivastava, N.; Swersky, K. Neural networks for machine learning lecture 6a overview of mini-batch gradient descent. Cited 2012, 14, 2. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef]
- Krueger, F.; Kreck, B.; Franke, A.; Andrews, S.R. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods 2012, 9, 145–151. [Google Scholar] [CrossRef]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Atkinson, S.R.; Marguerat, S.; Bähler, J. Exploring long non-coding RNAs through sequencing. Semin. Cell Dev. Biol. 2012, 23, 200–205. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Yost, R.A.; Enke, C. Tandem mass spectrometry. Science 1981, 214, 1135–1141. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid chromatography mass spectrometry-based proteomics: Biological and technological aspects. Ann. Appl. Stat. 2010, 4, 1797. [Google Scholar] [CrossRef]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef]
- Krone, N.; Hughes, B.A.; Lavery, G.G.; Stewart, P.M.; Arlt, W.; Shackleton, C.H. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J. Steroid Biochem. Mol. Biol. 2010, 121, 496–504. [Google Scholar] [CrossRef]
- Wang, M.; Wang, C.; Han, R.H.; Han, X. Novel advances in shotgun lipidomics for biology and medicine. Prog. Lipid Res. 2016, 61, 83–108. [Google Scholar] [CrossRef]
- Mardis, E.R. ChIP-seq: Welcome to the new frontier. Nat. Methods 2007, 4, 613–614. [Google Scholar] [CrossRef]
- Meers, M.P.; Bryson, T.D.; Henikoff, J.G.; Henikoff, S. Improved CUT&RUN chromatin profiling tools. eLife 2019, 8, e46314. [Google Scholar]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Tenenbaum, S.A.; Carson, C.C.; Lager, P.J.; Keene, J.D. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. USA 2000, 97, 14085–14090. [Google Scholar] [CrossRef]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef]
- Chen, R.; Yang, L.; Goodison, S.; Sun, Y. Deep-learning approach to identifying cancer subtypes using high-dimensional genomic data. Bioinformatics 2020, 36, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 2012, 7, e35236. [Google Scholar] [CrossRef] [PubMed]
- Witten, D.M.; Tibshirani, R. A framework for feature selection in clustering. J. Am. Stat. Assoc. 2010, 105, 713–726. [Google Scholar] [CrossRef]
- Tan, R.S.Y.C.; Ong, W.S.; Lee, K.H.; Lim, A.H.; Park, S.; Park, Y.H.; Lin, C.H.; Lu, Y.S.; Ono, M.; Ueno, T.; et al. HER2 expression, copy number variation and survival outcomes in HER2-low non-metastatic breast cancer: An international multicentre cohort study and TCGA-METABRIC analysis. BMC Med. 2022, 20, 105. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.; Fan, C.; Perou, C.M. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 135, 301–306. [Google Scholar] [CrossRef]
- Yang, H.; Chen, R.; Li, D.; Wang, Z. Subtype-GAN: A deep learning approach for integrative cancer subtyping of multi-omics data. Bioinformatics 2021, 37, 2231–2237. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, S. Deeply integrating latent consistent representations in high-noise multi-omics data for cancer subtyping. Brief. Bioinform. 2024, 25, bbae061. [Google Scholar] [CrossRef]
- He, K.; Fan, H.; Wu, Y.; Xie, S.; Girshick, R. Momentum contrast for unsupervised visual representation learning. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Seattle, WA, USA, 14–19 June 2020; pp. 9729–9738. [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Huang, H.; Zhu, L.; Huang, C.; Dong, Y.; Fan, L.; Tao, L.; Peng, Z.; Xiang, R. Identification of hub genes associated with clear cell renal cell carcinoma by integrated bioinformatics analysis. Front. Oncol. 2021, 11, 726655. [Google Scholar] [CrossRef] [PubMed]
- Sanjaya, P.; Maljanen, K.; Katainen, R.; Waszak, S.M.; Aaltonen, L.A.; Stegle, O.; Korbel, J.O.; Pitkänen, E. Mutation-Attention (MuAt): Deep representation learning of somatic mutations for tumour typing and subtyping. Genome Med. 2023, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Caulfield, M.; Davies, J.; Dennys, M.; Elbahy, L.; Fowler, T.; Hill, S.; Hubbard, T.; Jostins, L.; Maltby, N.; Mahon-Pearson, J.; et al. The National Genomics Research and Healthcare Knowledgebase. 2019. Available online: https://figshare.com/articles/dataset/GenomicEnglandProtocol_pdf/4530893/5?file=17384483 (accessed on 23 May 2025).
- Darmofal, M.; Suman, S.; Atwal, G.; Toomey, M.; Chen, J.F.; Chang, J.C.; Vakiani, E.; Varghese, A.M.; Balakrishnan Rema, A.; Syed, A.; et al. Deep-learning model for tumor-type prediction using targeted clinical genomic sequencing data. Cancer Discov. 2024, 14, 1064–1081. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Vibert, J.; Pierron, G.; Benoist, C.; Gruel, N.; Guillemot, D.; Vincent-Salomon, A.; Le Tourneau, C.; Livartowski, A.; Mariani, O.; Baulande, S.; et al. Identification of tissue of origin and guided therapeutic applications in cancers of unknown primary using deep learning and RNA sequencing (TransCUPtomics). J. Mol. Diagn. 2021, 23, 1380–1392. [Google Scholar] [CrossRef]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; de Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Maron, O.; Lozano-Pérez, T. A framework for multiple-instance learning. Adv. Neural Inf. Process. Syst. 1997, 10, 570–576. [Google Scholar]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef]
- Ostroverkhova, D.; Przytycka, T.M.; Panchenko, A.R. Cancer driver mutations: Predictions and reality. Trends Mol. Med. 2023, 29, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Ye, Z.; Qin, Y.; Xu, X.; Yu, X.; Zhuo, Q.; Ji, S. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Hou, J.P.; Ma, J. DawnRank: Discovering personalized driver genes in cancer. Genome Med. 2014, 6, 56. [Google Scholar] [CrossRef]
- Gu, H.; Xu, X.; Qin, P.; Wang, J. FI-net: Identification of cancer driver genes by using functional impact prediction neural network. Front. Genet. 2020, 11, 564839. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Chen, T.; Zhang, Y.; Xu, R.; Wang, W.; Cheng, M.; Chen, Q. Aberrant activation of hedgehog signalling promotes cell migration and invasion via matrix metalloproteinase-7 in ovarian cancer cells. J. Cancer 2019, 10, 990. [Google Scholar] [CrossRef]
- Trnski, D.; Gregorić, M.; Levanat, S.; Ozretić, P.; Rinčić, N.; Vidaković, T.M.; Kalafatić, D.; Maurac, I.; Orešković, S.; Sabol, M.; et al. Regulation of survivin isoform expression by GLI proteins in ovarian cancer. Cells 2019, 8, 128. [Google Scholar] [CrossRef]
- Borgquist, S.; Butt, T.; Almgren, P.; Shiffman, D.; Stocks, T.; Orho-Melander, M.; Manjer, J.; Melander, O. Apolipoproteins, lipids and risk of cancer. Int. J. Cancer 2016, 138, 2648–2656. [Google Scholar] [CrossRef]
- Lu, D.; Wang, J.; Shi, X.; Yue, B.; Hao, J. AHNAK2 is a potential prognostic biomarker in patients with PDAC. Oncotarget 2017, 8, 31775. [Google Scholar] [CrossRef] [PubMed]
- Talamillo, A.; Grande, L.; Ruiz-Ontañon, P.; Velásquez, C.; Mollinedo, P.; Torices, S.; Sanchez-Gomez, P.; Aznar, A.; Esparis-Ogando, A.; Lopez-Lopez, C.; et al. ODZ1 allows glioblastoma to sustain invasiveness through a Myc-dependent transcriptional upregulation of RhoA. Oncogene 2017, 36, 1733–1744. [Google Scholar] [CrossRef]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Bach, S.; Binder, A.; Montavon, G.; Klauschen, F.; Müller, K.R.; Samek, W. On pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS ONE 2015, 10, e0130140. [Google Scholar] [CrossRef]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. Deepwalk: Online learning of social representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; pp. 701–710. [Google Scholar]
- Peng, W.; Tang, Q.; Dai, W.; Chen, T. Improving cancer driver gene identification using multi-task learning on graph convolutional network. Brief. Bioinform. 2022, 23, bbab432. [Google Scholar] [CrossRef]
- He, M.; Wei, Z.; Wen, J.R. Convolutional neural networks on graphs with chebyshev approximation, revisited. Adv. Neural Inf. Process. Syst. 2022, 35, 7264–7276. [Google Scholar]
- Zhang, T.; Zhang, S.W.; Xie, M.Y.; Li, Y. A novel heterophilic graph diffusion convolutional network for identifying cancer driver genes. Brief. Bioinform. 2023, 24, bbad137. [Google Scholar] [CrossRef]
- Song, Q.; Li, M.; Li, Q.; Lu, X.; Song, K.; Zhang, Z.; Wei, J.; Zhang, L.; Wei, J.; Ye, Y.; et al. DeepAlloDriver: A deep learning-based strategy to predict cancer driver mutations. Nucleic Acids Res. 2023, 51, W129–W133. [Google Scholar] [CrossRef]
- Tomiya, A.; Nagai, Y. Equivariant transformer is all you need. arXiv 2023, arXiv:2310.13222. [Google Scholar]
- Shen, Q.; Cheng, F.; Song, H.; Lu, W.; Zhao, J.; An, X.; Liu, M.; Chen, G.; Zhao, Z.; Zhang, J. Proteome-scale investigation of protein allosteric regulation perturbed by somatic mutations in 7,000 cancer genomes. Am. J. Hum. Genet. 2017, 100, 5–20. [Google Scholar] [CrossRef]
- Tee, W.V.; Guarnera, E.; Berezovsky, I.N. On the allosteric effect of nsSNPs and the emerging importance of allosteric polymorphism. J. Mol. Biol. 2019, 431, 3933–3942. [Google Scholar] [CrossRef]
- Zeng, G.; Zhao, C.; Li, G.; Huang, Z.; Zhuang, J.; Liang, X.; Yu, X.; Fang, S. Identifying somatic driver mutations in cancer with a language model of the human genome. Comput. Struct. Biotechnol. J. 2025, 27, 531–540. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, L.; Zhang, K.; Zhang, Y.; Hu, S.; Zhao, T.; Teng, H.; Li, X.; Jiang, Y.; Ji, L.; et al. VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018, 46, D1039–D1048. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilic, N.; Shrestha, Y.; Zou, L.; Kamburov, A.; Zhu, C.; Yang, X.; Lubonja, R.; Tran, N.; Nguyen, C.; et al. Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 2016, 6, 714–726. [Google Scholar] [CrossRef]
- Martelotto, L.G.; Ng, C.K.; De Filippo, M.R.; Zhang, Y.; Piscuoglio, S.; Lim, R.S.; Shen, R.; Norton, L.; Reis-Filho, J.S.; Weigelt, B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014, 15, 484. [Google Scholar] [CrossRef]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Zhao, L.; Dong, Q.; Luo, C.; Wu, Y.; Bu, D.; Qi, X.; Luo, Y.; Zhao, Y. DeepOmix: A scalable and interpretable multi-omics deep learning framework and application in cancer survival analysis. Comput. Struct. Biotechnol. J. 2021, 19, 2719–2725. [Google Scholar] [CrossRef]
- Hornung, R.; Wright, M.N. Block forests: Random forests for blocks of clinical and omics covariate data. BMC Bioinform. 2019, 20, 358. [Google Scholar] [CrossRef]
- Lee, C.; Zame, W.; Yoon, J.; Van Der Schaar, M. Deephit: A deep learning approach to survival analysis with competing risks. In Proceedings of the AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018; Volume 32. [Google Scholar]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.; Li, L. MMOSurv: Meta-learning for few-shot survival analysis with multi-omics data. Bioinformatics 2025, 41, btae684. [Google Scholar] [CrossRef]
- Nichol, A.; Schulman, J. Reptile: A scalable metalearning algorithm. arXiv 2018, arXiv:1803.02999. [Google Scholar]
- Kalakoti, Y.; Yadav, S.; Sundar, D. SurvCNN: A discrete time-to-event cancer survival estimation framework using image representations of omics data. Cancers 2021, 13, 3106. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, C.; Li, X.; Dubrawski, A. Deep survival machines: Fully parametric survival regression and representation learning for censored data with competing risks. IEEE J. Biomed. Health Inform. 2021, 25, 3163–3175. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Du, Y.; Li, S.; Ma, S.; Jiang, Y.; Wu, C. Robust network-based regularization and variable selection for high-dimensional genomic data in cancer prognosis. Genet. Epidemiol. 2019, 43, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, C.; Yang, Z.; Zhou, M.; Sun, J. Multi-omics deep-learning prediction of homologous recombination deficiency-like phenotype improved risk stratification and guided therapeutic decisions in gynecological cancers. IEEE J. Biomed. Health Inform. 2023, 29, 1861–1871. [Google Scholar] [CrossRef]
- Chen, R.J.; Lu, M.Y.; Williamson, D.F.; Chen, T.Y.; Lipkova, J.; Noor, Z.; Shaban, M.; Shady, M.; Williams, M.; Joo, B.; et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell 2022, 40, 865–878. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, C.; Bai, Y.; Liu, A.; Gong, Y.; Wang, Y.P.; Deng, H.W. Autosurv: Interpretable deep learning framework for cancer survival analysis incorporating clinical and multi-omics data. NPJ Precis. Oncol. 2024, 8, 4. [Google Scholar] [CrossRef]
- Desmedt, C.; Piette, F.; Loi, S.; Wang, Y.; Lallemand, F.; Haibe-Kains, B.; Viale, G.; Delorenzi, M.; Zhang, Y.; d’Assignies, M.S. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res. 2007, 13, 3207–3214. [Google Scholar] [CrossRef]
- Rollo, C.; Pancotti, C.; Sartori, F.; Caranzano, I.; D’Amico, S.; Carota, L.; Casadei, F.; Birolo, G.; Lanino, L.; Sauta, E.; et al. VAE-Surv: A novel approach for genetic-based clustering and prognosis prediction in myelodysplastic syndromes. Comput. Methods Programs Biomed. 2025, 261, 108605. [Google Scholar] [CrossRef]
- Chapfuwa, P.; Li, C.; Mehta, N.; Carin, L.; Henao, R. Survival cluster analysis. In Proceedings of the ACM Conference on Health, Inference, and Learning, Toronto, ON, Canada, 2–4 April 2020; pp. 60–68. [Google Scholar]
- Yang, Z.; Wei, T.; Liang, Y.; Yuan, X.; Gao, R.; Xia, Y.; Zhou, J.; Zhang, Y.; Yu, Z. A foundation model for generalizable cancer diagnosis and survival prediction from histopathological images. Nat. Commun. 2025, 16, 2366. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.; Brettin, T.S.; Zhu, Y.; Narykov, O.; Clyde, A.; Overbeek, J.; Stevens, R.L. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front. Med. 2023, 10, 1086097. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Z.; Jiang, R.; Zhou, M. DeepCDR: A hybrid graph convolutional network for predicting cancer drug response. Bioinformatics 2020, 36, i911–i918. [Google Scholar] [CrossRef]
- Nguyen, T.; Nguyen, G.T.; Nguyen, T.; Le, D.H. Graph convolutional networks for drug response prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 146–154. [Google Scholar] [CrossRef]
- Liu, X.; Song, C.; Huang, F.; Fu, H.; Xiao, W.; Zhang, W. GraphCDR: A graph neural network method with contrastive learning for cancer drug response prediction. Brief. Bioinform. 2022, 23, bbab457. [Google Scholar] [CrossRef]
- Jiang, L.; Jiang, C.; Yu, X.; Fu, R.; Jin, S.; Liu, X. DeepTTA: A transformer-based model for predicting cancer drug response. Brief. Bioinform. 2022, 23, bbac100. [Google Scholar] [CrossRef]
- Wang, H.; Dai, C.; Wen, Y.; Wang, X.; Liu, W.; He, S.; Bo, X.; Peng, S. GADRP: Graph convolutional networks and autoencoders for cancer drug response prediction. Brief. Bioinform. 2023, 24, bbac501. [Google Scholar] [CrossRef]
- Liu, H.; Peng, W.; Dai, W.; Lin, J.; Fu, X.; Liu, L.; Liu, L.; Yu, N. Improving anti-cancer drug response prediction using multi-task learning on graph convolutional networks. Methods 2024, 222, 41–50. [Google Scholar] [CrossRef]
- Jang, I.S.; Neto, E.C.; Guinney, J.; Friend, S.H.; Margolin, A.A. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. In Proceedings of the Biocomputing 2014, Kohala Coast, HI, USA, 3–7 January 2014; pp. 63–74. [Google Scholar]
- Pozdeyev, N.; Yoo, M.; Mackie, R.; Schweppe, R.E.; Tan, A.C.; Haugen, B.R. Integrating heterogeneous drug sensitivity data from cancer pharmacogenomic studies. Oncotarget 2016, 7, 51619. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Codicè, F.; Pancotti, C.; Rollo, C.; Moreau, Y.; Fariselli, P.; Raimondi, D. The specification game: Rethinking the evaluation of drug response prediction for precision oncology. J. Cheminform. 2025, 17, 33. [Google Scholar] [CrossRef]
- Baptista, D.; Ferreira, P.G.; Rocha, M. Deep learning for drug response prediction in cancer. Brief. Bioinform. 2021, 22, 360–379. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, K.; Born, J.; Chouvardas, P.; Rapsomaniki, M.; Kruithof-de Julio, M. Overcoming limitations in current measures of drug response may enable AI-driven precision oncology. NPJ Precis. Oncol. 2024, 8, 95. [Google Scholar] [CrossRef]
- Szalai, B.; Gáspár, I.; Kaszás, V.; Mérő, L.; Sztilkovics, M.; Szalay, K.Z. The EFFECT benchmark suite: Measuring cancer sensitivity prediction performance-without the bias. bioRxiv 2023, 10, 1–25. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Architecture | Task | Data Type | Strength | Possible Limitations | Datasets |
|---|---|---|---|---|---|---|
| DeepType (https://github.com/runpuchen/DeepType, accessed on 23 May 2025) [87] | FFNN | Tumor subtyping | GEx | Supervised classification with K-means clustering | Single omics data. No. of cancer types | METABRIC (BRCA), TCGA (BLCA) |
| Subtype-GAN (https://github.com/haiyang1986/Subtype-GAN, accessed on 23 May 2025) [93] | GAN | Tumor subtyping | CNVs, DNAm, miRNA, mRNA | Multiomics data. Adversarial learning with GMM clustering | High-dimensional data. No of cancer types | TCGA (10 cancer types) |
| DICLR (https://github.com/ykxhs/DILCR, accessed on 23 May 2025) [95] | Custom VAE | Tumor subtyping | DNAm, miRNA, mRNA | Multiomics data. Noise disentanglement. Self-supervised clustering | High-dimensional data. No. of cancer types | TCGA (10 cancer types) |
| MuAt (https://github.com/primasanjaya/mutation-attention, accessed on 23 May 2025) [99] | TRNN | Tumor typing, subtyping, and CUP | SNVs/MNVs, indels, SVs | Many mutational events. Attention mechanism. No. of cancer types | High-dimensional data. Single omics | PCAWG, TCGA (24 cancer types) |
| GDD-ENS (https://github.com/mmdarmofal/GDD_ENS, accessed on 23 May 2025) [102] | FFNN | Tumor typing and CUP | Targeted sequencing | Ensemble neural network. Gene panel | Standard neural network architecture | MSK-IMPACT (38 cancer types) |
| TransCUP-tomics [104] | VAE + RF | CUP | GEx | No. of cancer types | Single omics. No code available. | TCGA (39 cancer types), GTEx, HPA |
| DeepTumour (https://github.com/ICGC-TCGA-PanCancer/TumorType-WGS, accessed on 23 May 2025) [105] | FFNN | CUP | SNVs, Indels, CNVs, SVs | Many mutational events. Passenger mutations. No. of cancer types | Single omics | PCAWG (24 cancer types), HMF |
| TOAD (https://github.com/mahmoodlab/TOAD, accessed on 23 May 2025) [32] | CNN encoder + attention modules | CUP and tumor status | H&E images | Weakly supervised instance learning. Attention mechanism. Multitask learning | Single omics | BWH (18 cancer types), TCGA |
| Model | Architecture | Task | Data Type | Strength | Possible Limitations | Datasets |
|---|---|---|---|---|---|---|
| FI-Net [120] | FFNN | Cancer drivers | Mutations, DNAm, GEx, HiC | FIS-score. No. of cancer types. Multiomics data. | High-dimensional data, standard neural network architecture. Not evaluated on an independent test set of genes | TCGA (31 cancer types), NCG, CGC |
| EMOGI (https://github.com/schulter/EMOGI, accessed on 23 May 2025) [127] | GCNs | Cancer drivers | Mutations, DNAm, GEx, PPI network | LRP for explainability. Multi-omics data | High-dimensional data | TCGA (16 cancer types), NCG |
| MTGCN (https://github.com/haiyang1986/Subtype-GAN, accessed on 23 May 2025) [131] | GCN | Cancer drivers and link prediction in PPI network | Mutations, DNAm, GEx, PPI network | Chebyshev GCN. Multi-omics data. Multitask learning | High-dimensional data | TCGA, NCG, KEGG, OncoKB |
| HGDC (https://github.com/NWPU-903PR/HGDC, accessed on 23 May 2025) [133] | Diffusion GCN with PPR | Cancer drivers | Mutations, DNAm, GEx, PPI, and GGI networks | Diffusion process. Multi-omics data. Can deal with heterophilic networks | High-dimensional data | TCGA, KEGG, Reactom, GGNet, PPNet |
| DeepAlloDriver (https://mdl.shsmu.edu.cn/DeepAlloDriver, accessed on 23 May 2025) [134] | EGNN | Cancer drivers on allosteric sites | Gene symbol and amino acid substitution | Equivariant architecture. Attention mechanism. Prediction on allosteric sites. Web Server | - | RCSB Protein Data Bank, Allosteric Database |
| GenomeBert (https://github.com/GaryinDeep/GenomeBert, accessed on 23 May 2025) [138] | LLM + XGB | Oncogene prediction, tumor suppressor genes and cancer drivers | DNA sequences | LLM for feature extraction | Interpretability. Computationally expensive. | GRC, CGC, OncoKB, FASMIC |
| Model | Architecture | Task | Data Type | Strength | Possible Limitations | Datasets |
|---|---|---|---|---|---|---|
| DeepOmix [145] | FFNN | Risk prediction and patient stratification | CNV, DNAm, GEx, Mutations | Functional module layer and cox proportional hazard layer | No code available | TCGA |
| MMOSurv (https://github.com/LiminLi-xjtu/MMOSurv, accessed on 23 May 2025) [149] | FFNN | Risk prediction | GEx, miRNA | Meta-learning for few-shot learning | No external validation dataset | TCGA |
| MODeepHRD (https://github.com/ZhouSunLab-Workshops/MODeepHRD, accessed on 23 May 2025) [154] | Convolutional Attention Mechanism | HRD classification and prognosis prediction | miRNA, DNAm, RNA-seq, mutations | GAN-based augmentation. Survival based on the HRD status | Synthetic data can lead to bias | ArrayExpress, ICGC, TCGA, GEO, SCAN-B |
| PORPOISE (https://github.com/mahmoodlab/PORPOISE, accessed on 23 May 2025) [155] | MIL + FFNN | Time bin classification | WSIs, mutations, CNV, RNA-seq | Multi-omics data. Explainability. WSI images. | Prediction of time bins. Foundation model based on ResNet-50 (obsolete) | TCGA |
| AUTOSurv (https://github.com/jianglindong93/AUTOSurv, accessed on 23 May 2025) [156] | VAE + FFNN | Prognostic index | CL, miRNA, GEx | VAE and CL | High-dimensional data | Caldas-BC, ICGC, TCGA |
| VAE-Surv (https://github.com/compbiomed-unito/VAE-Surv, accessed on 23 May 2025) [158] | VAE + DeepSurv | Risk prediction and patient stratification | CL, Mutations, cytogenetic alterations | Unified, genetic patient clustering and survival prediction | Tested only on a specific use case | GenoMed4All, IWG-PM MDS cohorts |
| ClamSurvival (https://github.com/Zhcyoung/BEPH, accessed on 23 May 2025) [160] | Attention + FFNN | Risk prediction | WSIs, CL | Multitask learning. WSI images. | - | TCGA, BreakHis, LC25000, NCT-CRC-HE-100K, CAMELYON16, BACH |
| Model | Architecture | Task | Data Type | Strength | Possible Limitations | Datasets |
|---|---|---|---|---|---|---|
| DeepCDR (https://github.com/kimmo1019/DeepCDR, accessed on 23 May 2025) [166] | CNN + GCN | IC50 regression and DS classification | SNPs, GEx, DNAm, DMG | Multi-omics integration; validation on TCGA | - | GDSC, CCLE, TCGA |
| GraphDRP (https://github.com/hauldhut/GraphDRP, accessed on 23 May 2025) [167] | CNN + GNN | IC50 regression | SNPs, CNV, DMG | Comparative GNN evaluation; interpretable via saliency maps | Limited to binary genomic features; single dataset | GDSC |
| GraphCDR (https://github.com/hauldhut/GraphDRP, accessed on 23 May 2025) [168] | GNN + contrastive learning | DS classification | GEx, SNPs, DNAm, DMG | Heterogeneous graph; contrastive learning; multi-omics integration | - | GDSC, CCLE |
| DeepTTA (https://github.com/CZenkert/DeepTTA-implementation, accessed on 23 May 2025) [169] | Transformer + FFNN | IC50 regression and DS classification | GEx, Drug SMILES substructures (ESPF) | Transformer-based drug encoding; interpretable substructures | Only GEx omics data; single dataset | GDSC |
| GADRP (https://github.com/flora619/GADRP, accessed on 23 May 2025) [170] | Autoencoder + ILGCN | IC50 regression | GEx, DNAm, CNV, miRNA, DFPs | Residual GCN; multi-omics; PRISM data | - | PRISM, CCLE |
| MTIGCN (https://github.com/weiba/MTIGCN, accessed on 23 May 2025) [171] | GNN + MTL | IC50 regression and DS classification | GEx, DFPs | Multi-task learning; in vivo evaluation | No drug structure modeling | GDSC, CCLE, PDX, TCGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sartori, F.; Codicè, F.; Caranzano, I.; Rollo, C.; Birolo, G.; Fariselli, P.; Pancotti, C. A Comprehensive Review of Deep Learning Applications with Multi-Omics Data in Cancer Research. Genes 2025, 16, 648. https://doi.org/10.3390/genes16060648
Sartori F, Codicè F, Caranzano I, Rollo C, Birolo G, Fariselli P, Pancotti C. A Comprehensive Review of Deep Learning Applications with Multi-Omics Data in Cancer Research. Genes. 2025; 16(6):648. https://doi.org/10.3390/genes16060648
Chicago/Turabian StyleSartori, Flavio, Francesco Codicè, Isabella Caranzano, Cesare Rollo, Giovanni Birolo, Piero Fariselli, and Corrado Pancotti. 2025. "A Comprehensive Review of Deep Learning Applications with Multi-Omics Data in Cancer Research" Genes 16, no. 6: 648. https://doi.org/10.3390/genes16060648
APA StyleSartori, F., Codicè, F., Caranzano, I., Rollo, C., Birolo, G., Fariselli, P., & Pancotti, C. (2025). A Comprehensive Review of Deep Learning Applications with Multi-Omics Data in Cancer Research. Genes, 16(6), 648. https://doi.org/10.3390/genes16060648