
Exploring the Impact of Mitonuclear Discordance on Disease in Latin American Admixed Populations

Abstract
1. Introduction
1.1. Mitochondrial Function Depends on Nuclear and Mitochondrial Factors
1.2. Mitonuclear Discordance Leads to Dysfunctional Mitochondria
1.3. Admixture Leads to Mitonuclear Discordance in Humans
1.4. The Effect of Mitonuclear Discordance Due to Admixture Is Unclear
1.5. Mitonuclear Interactions Can Modulate Disease Phenotypes
2. Materials and Methods
2.1. Datasets
2.2. Imputation of Exome Data
2.3. Ancestry Inference
2.4. Haplogroup Assignment
2.5. Mitonuclear Discordance
2.6. Gene Sets
2.7. Statistics
3. Results
3.1. Imputation
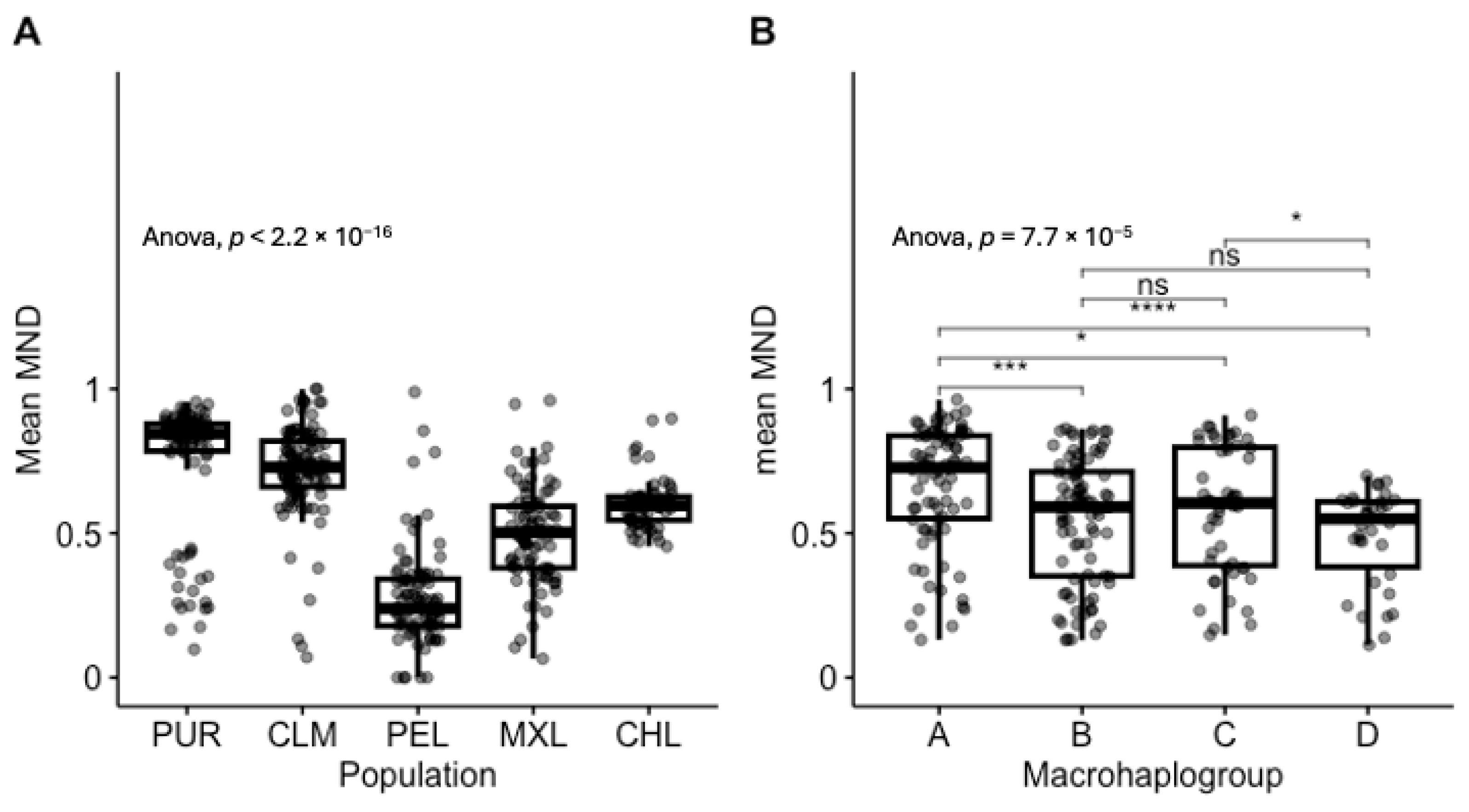
3.2. MND in Healthy Individuals Shows Strong Population and Haplogroup-Specific Distributions
3.3. Affected Individuals Have Lower MND
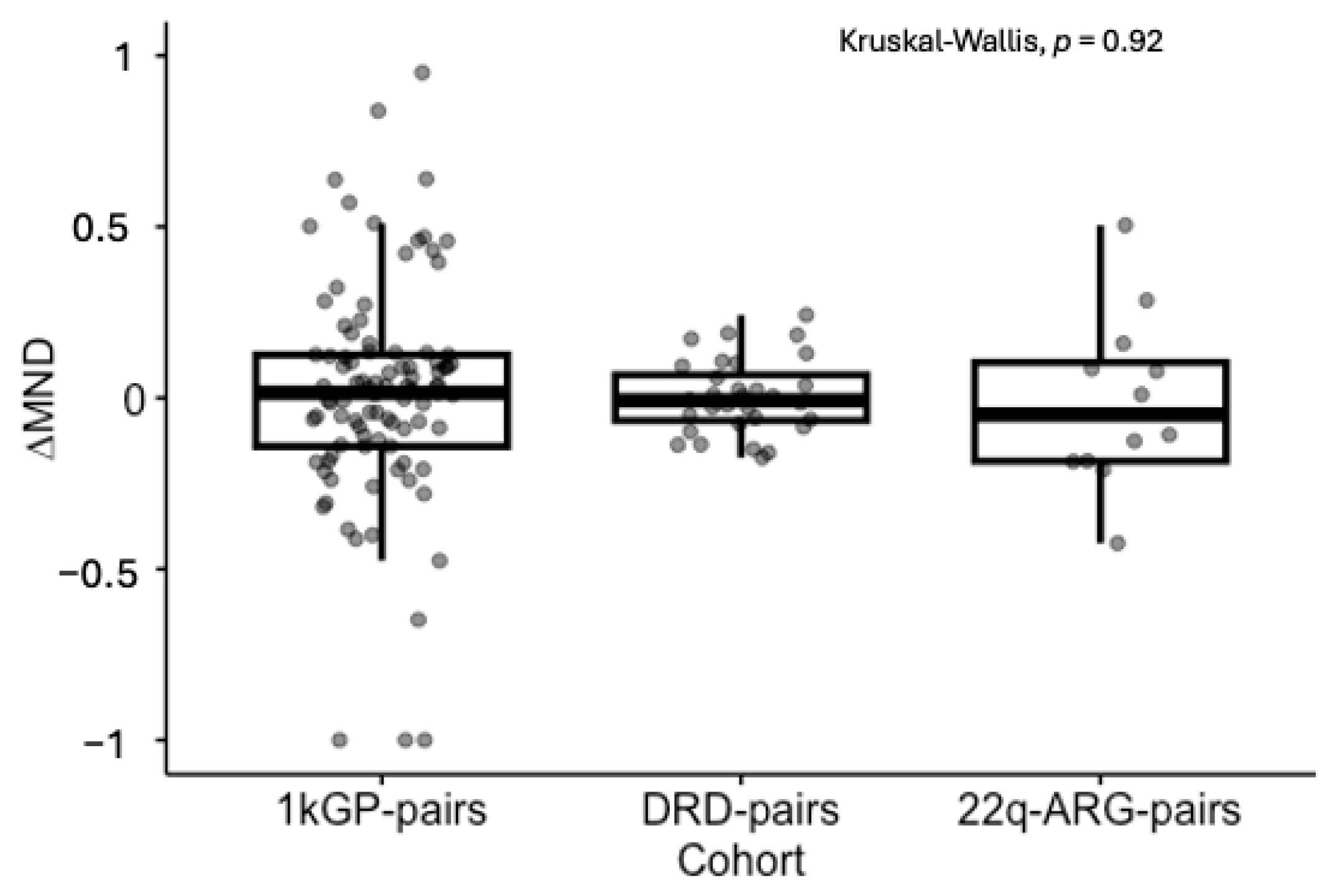
3.4. Signs of Constraint of ∆MND
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFR | African |
| AMR | Admixed American |
| ANOVA | Analysis of Variance |
| CN | Copy Number |
| DECIPHERD | Decoding Complex Inherited Phenotypes in Rare Disorders |
| DRD | Decoding Rare Disorders |
| EUR | European |
| GWAS | Genome-Wide Association Study |
| MND | Mitonuclear Discordance |
| MRT | Mitochondrial Replacement Therapy |
| NAT | Native American |
| mtDNA | Mitochondrial DNA |
| OXPHOS | Oxidative Phosphorylation |
| SNP | Single-Nucleotide Polymorphism |
| YBP | Years Before Present |
References
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Zachar, I.; Boza, G. Endosymbiosis before eukaryotes: Mitochondrial establishment in protoeukaryotes. Cell. Mol. Life Sci. 2020, 77, 3503–3523. [Google Scholar] [CrossRef]
- Levin, L.; Blumberg, A.; Barshad, G.; Mishmar, D. Mito-nuclear co-evolution: The positive and negative sides of functional ancient mutations. Front. Genet. 2014, 5, 448. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2020, 22, 106–118. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2020, 22, 54–70. [Google Scholar] [CrossRef]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Churchman, L.S. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Gutierrez, N.M.; Morey, R.; Van Dyken, C.; Kang, E.; Hayama, T.; Lee, Y.; Li, Y.; Tippner-Hedges, R.; Wolf, D.P.; et al. Incompatibility between Nuclear and Mitochondrial Genomes Contributes to an Interspecies Reproductive Barrier. Cell Metab. 2016, 24, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Mossman, J.A.; Tross, J.G.; Jourjine, N.A.; Li, N.; Wu, Z.; Rand, D.M. Mitonuclear Interactions Mediate Transcriptional Responses to Hypoxia in Drosophila. Mol. Biol. Evol. 2016, 34, 447–466. [Google Scholar] [CrossRef]
- Nguyen, T.H.M.; Sondhi, S.; Ziesel, A.; Paliwal, S.; Fiumera, H.L. Mitochondrial-nuclear coadaptation revealed through mtDNA replacements in Saccharomyces cerevisiae. BMC Evol. Biol. 2020, 20, 128. [Google Scholar] [CrossRef]
- Ellison, C.K.; Burton, R.S. Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution 2008, 62, 631–638. [Google Scholar] [CrossRef]
- Morales, H.E.; Pavlova, A.; Amos, N.; Major, R.; Kilian, A.; Greening, C.; Sunnucks, P. Concordant divergence of mitogenomes and a mitonuclear gene cluster in bird lineages inhabiting different climates. Nat. Ecol. Evol. 2018, 2, 1258–1267. [Google Scholar] [CrossRef]
- Baris, T.Z.; Wagner, D.N.; Dayan, D.I.; Du, X.; Blier, P.U.; Pichaud, N.; Oleksiak, M.F.; Crawford, D.L. Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genet. 2017, 13, e1006517. [Google Scholar] [CrossRef]
- Rishishwar, L.; Jordan, I.K. Implications of human evolution and admixture for mitochondrial replacement therapy. BMC Genom. 2017, 18, 140. [Google Scholar] [CrossRef]
- Zaidi, A.A.; Makova, K.D. Investigating mitonuclear interactions in human admixed populations. Nat. Ecol. Evol. 2019, 3, 213–222. [Google Scholar] [CrossRef]
- Rebolledo-Jaramillo, B.; Su, M.S.-W.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.T.; Wang, L.; Conley, A.B.; Rishishwar, L.; Mariño-Ramírez, L.; Valderrama-Aguirre, A.; Jordan, I.K. Genetic ancestry, admixture and health determinants in Latin America. BMC Genom. 2018, 19, 75–87. [Google Scholar] [CrossRef]
- Jordan, I.K. The Columbian Exchange as a source of adaptive introgression in human populations. Biol. Direct 2016, 11, 17. [Google Scholar] [CrossRef]
- Ruiz-Linares, A.; Adhikari, K.; Acuña-Alonzo, V.; Quinto-Sanchez, M.; Jaramillo, C.; Arias, W.; Fuentes, M.; Pizarro, M.; Everardo, P.; de Avila, F.; et al. Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals. PLOS Genet. 2014, 10, e1004572. [Google Scholar] [CrossRef]
- Greenfield, A.; Braude, P.; Flinter, F.; Lovell-Badge, R.; Ogilvie, C.; Perry, A.C.F. Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat. Biotechnol. 2017, 35, 1059–1068. [Google Scholar] [CrossRef]
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, 216.e7–216.e10. [Google Scholar] [CrossRef]
- Lee, H.; Song, J.; Shin, C.; Park, D.; Park, K.; Lee, K.; Koh, C.-S. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pr. 1998, 42, 161–167. [Google Scholar] [CrossRef]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef]
- Hill, G.E.; Havird, J.C.; Sloan, D.B.; Burton, R.S.; Greening, C.; Dowling, D.K. Assessing the fitness consequences of mitonuclear interactions in natural populations. Biol. Rev. 2018, 94, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Tuna, S.; Keogh, M.J.; Smith, G.; Aitman, T.J.; Beales, P.L.; Bennett, D.L.; Gale, D.P.; Bitner-Glindzicz, M.A.K.; Black, G.C.; et al. Germline selection shapes human mitochondrial DNA diversity. Science 2019, 364, 6520. [Google Scholar] [CrossRef]
- Ludwig-Słomczyńska, A.H.; Seweryn, M.T.; Kapusta, P.; Pitera, E.; Handelman, S.K.; Mantaj, U.; Cyganek, K.; Gutaj, P.; Dobrucka, Ł.; Wender-Ożegowska, E.; et al. Mitochondrial GWAS and association of nuclear—Mitochondrial epistasis with BMI in T1DM patients. BMC Med. Genom. 2020, 13, 97. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Patterson, C.; McFall, G.P.; Gross, A.; Michaelis, E.K.; Goate, A.; Swerdlow, R.H.; Pa, J. Mitonuclear interactions influence Alzheimer’s disease risk. Neurobiol. Aging 2019, 87, 138.e7–138.e14. [Google Scholar] [CrossRef]
- Schulmann, A.; Ryu, E.; Goncalves, V.; Rollins, B.; Christiansen, M.; Frye, M.A.; Biernacka, J.; Vawter, M.P. Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Mol. Neuropsychiatry 2019, 5, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gershoni, M.; Levin, L.; Ovadia, O.; Toiw, Y.; Shani, N.; Dadon, S.; Barzilai, N.; Bergman, A.; Atzmon, G.; Wainstein, J.; et al. Disrupting mitochondrial-nuclear coevolution affects OXPHOS complex i integrity and impacts human health. Genome Biol. Evol. 2014, 6, 2665–2680. [Google Scholar] [CrossRef]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D. 22q11.21 deletion syndromes: A review of proximal, central, and distal deletions and their associated features. Cytogenet. Genome Res. 2015, 146, 89–99. [Google Scholar] [CrossRef]
- Mcdonald-Mcginn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; A Driscoll, D.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet. Med. 2001, 3, 23–29. [Google Scholar] [CrossRef]
- Cancrini, C.; Puliafito, P.; Digilio, M.C.; Soresina, A.; Martino, S.; Rondelli, R.; Consolini, R.; Ruga, E.M.; Cardinale, F.; Finocchi, A.; et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J. Pediatr. 2014, 164, 1475–1480.e2. [Google Scholar] [CrossRef]
- Devaraju, P.; Zakharenko, S.S. Mitochondria in complex psychiatric disorders: Lessons from mouse models of 22q11.2 deletion syndrome. BioEssays 2017, 39, 1600177. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ryan, S.K.; Deboer, E.; Cook, K.; Fitzgerald, S.; Lachman, H.M.; Wallace, D.C.; Goldberg, E.M.; Anderson, S.A. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry 2019, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef]
- Poli, M.C.; Rebolledo-Jaramillo, B.; Lagos, C.; Orellana, J.; Moreno, G.; Martín, L.M.; Encina, G.; Böhme, D.; Faundes, V.; Zavala, M.J.; et al. Decoding complex inherited phenotypes in rare disorders: The DECIPHERD initiative for rare undiagnosed diseases in Chile. Eur. J. Hum. Genet. 2024, 32, 1227–1237. [Google Scholar] [CrossRef]
- Rebolledo-Jaramillo, B.; Obregon, M.G.; Huckstadt, V.; Gomez, A.; Repetto, G.M. Contribution of mitochondrial DNA heteroplasmy to the congenital cardiac and palatal phenotypic variability in maternally transmitted 22q11.2 deletion syndrome. Genes 2021, 12, 92. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef]
- Browning, S.R.; Waples, R.K.; Browning, B.L. Fast, accurate local ancestry inference with FLARE. Am. J. Hum. Genet. 2023, 110, 326–335. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Fields, P.D.; Havird, J.C. Mitonuclear linkage disequilibrium in human populations. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151704. [Google Scholar] [CrossRef]
- Torres-Gonzalez, E.; Makova, K.D. Exploring the Effects of Mitonuclear Interactions on Mitochondrial DNA Gene Expression in Humans. Front. Genet. 2022, 13, 797129. [Google Scholar] [CrossRef]
- Lake, N.J.; Ma, K.; Liu, W.; Battle, S.L.; Laricchia, K.M.; Tiao, G.; Puiu, D.; Ng, K.K.; Cohen, J.; Compton, A.G.; et al. Quantifying constraint in the human mitochondrial genome. Nature 2024, 635, 390–397. [Google Scholar] [CrossRef]
- Schaefer, P.M.; Alves, L.S.; Lvova, M.; Huang, J.; Rathi, K.; Janssen, K.; Butic, A.; Yardeni, T.; Morrow, R.; Lott, M.; et al. Combination of common mtDNA variants results in mitochondrial dysfunction and a connective tissue dysregulation. Proc. Natl. Acad. Sci. USA 2022, 119, e2212417119. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S. The Role of Mitonuclear Incompatibility in Bipolar Disorder Susceptibility and Resilience Against Environmental Stressors. Front. Genet. 2021, 12, 636294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xiang, F.; Li, D.; Yang, F.; Yu, S.; Wang, X. Adult-onset combined oxidative phosphorylation deficiency type 14 manifests as epileptic status: A new phenotype and literature review. BMC Neurol. 2024, 24, 15. [Google Scholar] [CrossRef]
- Burton, R.S. The role of mitonuclear incompatibilities in allopatric speciation. Cell. Mol. Life Sci. 2022, 79, 103. [Google Scholar] [CrossRef]
- Honorato-Mauer, J.; Shah, N.N.; Maihofer, A.X.; Zai, C.C.; Belangero, S.; Nievergelt, C.M.; Santoro, M.; Atkinson, E.G. Characterizing features affecting local ancestry inference performance in admixed populations. Am. J. Hum. Genet. 2025, 112, 224–234. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Dataset Origin | Short Name | # of Samples | # Pairs/ Trios | Usage |
|---|---|---|---|---|
| 1000 Genomes Project Phase 3 | 1kGP-ALL | 3202 | 602 | To determine AMR, EUR, and AFR haplotype references |
| 1000 Genomes Project Phase 3 | 1kGP-AMR | 385 | 98 | To describe MND and ∆MND in healthy Latin American individuals |
| DECIPHERD | DRD-healthy | 63 | 0 | To describe MND in healthy Chilean individuals |
| DECIPHERD | DRD-affected | 75 | 0 | To describe MND in affected Chilean individuals |
| DECIPHERD | DRD-pairs | 64 | 32 | To describe ∆MND in affected Chilean mother–offspring pairs |
| 22q11.2DS—Chile | 22q-CHL | 58 | 0 | To describe MND in a known disease |
| 22q11.2DS—Argentina | 22q-ARG | 26 | 13 | To describe MND and ∆MND in a known disease |
| All Mitonuclear Genes | OXPHOS Genes | ||||||
|---|---|---|---|---|---|---|---|
| N | Mean | sd | Cohen’s d | Mean | sd | Cohen’s d | |
| DRD-healthy | 63 | 0.6 | 0.09 | — | 0.62 | 0.1 | — |
| DRD-affected | 75 | 0.56 | 0.12 | 0.38 | 0.55 | 0.13 | 0.76 |
| 22q-CHL | 58 | 0.57 | 0.13 | 0.27 | 0.57 | 0.13 | 0.54 |
| 22q-ARG | 26 | 0.43 | 0.24 | 0.94 | 0.43 | 0.25 | 1.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz, M.; Böhme, D.; Repetto, G.M.; Rebolledo-Jaramillo, B. Exploring the Impact of Mitonuclear Discordance on Disease in Latin American Admixed Populations. Genes 2025, 16, 638. https://doi.org/10.3390/genes16060638
Ruiz M, Böhme D, Repetto GM, Rebolledo-Jaramillo B. Exploring the Impact of Mitonuclear Discordance on Disease in Latin American Admixed Populations. Genes. 2025; 16(6):638. https://doi.org/10.3390/genes16060638
Chicago/Turabian StyleRuiz, Mauricio, Daniela Böhme, Gabriela M. Repetto, and Boris Rebolledo-Jaramillo. 2025. "Exploring the Impact of Mitonuclear Discordance on Disease in Latin American Admixed Populations" Genes 16, no. 6: 638. https://doi.org/10.3390/genes16060638
APA StyleRuiz, M., Böhme, D., Repetto, G. M., & Rebolledo-Jaramillo, B. (2025). Exploring the Impact of Mitonuclear Discordance on Disease in Latin American Admixed Populations. Genes, 16(6), 638. https://doi.org/10.3390/genes16060638