
Phase Determination and Demonstration of Parental Mosaicism of Intragenic PRKN Deletions Initially Identified by Chromosomal Microarray Analysis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chromosomal Microarray
2.2. Long-Range PCR and Sanger Sequencing
- Hold: 94 °C for 1 min
- Denature: 95 °C for 15 min
- Anneal: 65 °C for 30 min
- Extend: 68 °C for 15 min
- Steps 2–4, 35 cycles
- Final extension: 72 °C for 10 min
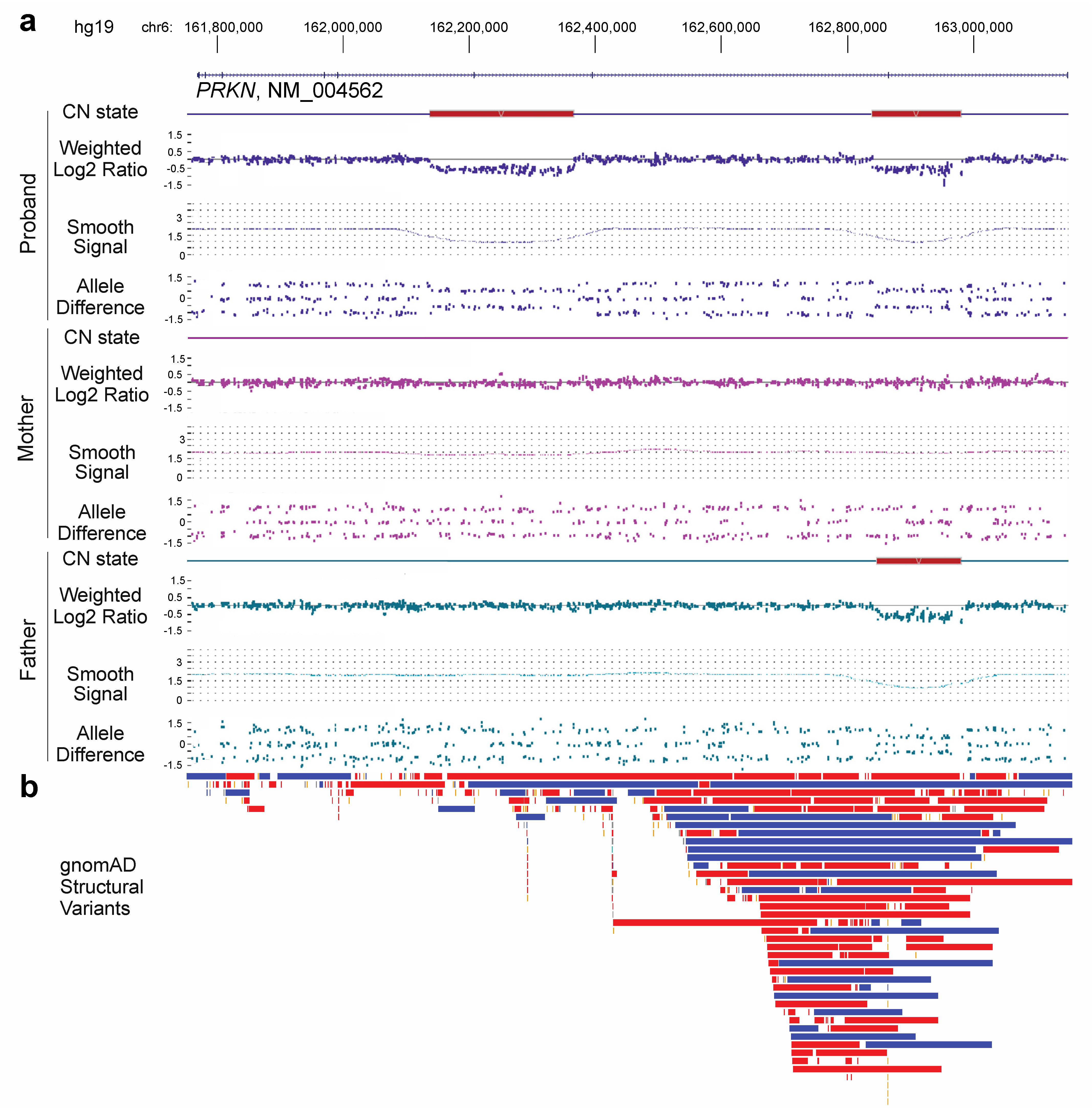
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willis, A.W.; Roberts, E.; Beck, J.C.; Fiske, B.; Ross, W.; Savica, R.; Eeden, S.K.V.D.; Tanner, C.M.; Marras, C.; Alcalay, R.; et al. Incidence of Parkinson disease in North America. npj Parkinsons Disease 2022, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Doherty, K.M.; Silveira-Moriyama, L.; Parkkinen, L.; Healy, D.G.; Farrell, M.; Mencacci, N.E.; Ahmed, Z.; Brett, F.M.; Hardy, J.; Quinn, N.; et al. Parkin disease: A clinicopathologic entity? JAMA Neurol. 2013, 70, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.-I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Chung, K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef]
- Abbas, N.; Lücking, C.B.; Ricard, S.; Dürr, A.; Bonifati, V.; De Michele, G.; Bouley, S.; Vaughan, J.R.; Gasser, T.; Marconi, R.; et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum. Mol. Genet. 1999, 8, 567–574. [Google Scholar] [CrossRef]
- Clausen, L.; Okarmus, J.; Voutsinos, V.; Meyer, M.; Lindorff-Larsen, K.; Hartmann-Petersen, R. PRKN-linked familial Parkinson’s disease: Cellular and molecular mechanisms of disease-linked variants. Cell. Mol. Life Sci. 2024, 81, 223. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, S.J.; I Bustos, B.; Hu, J.; Krainc, D.; Joseph, T.; Hehir, J.; Tan, M.; Zhang, W.; Escott-Price, V.; Williams, N.M.; et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. Hum. Mol. Genet. 2021, 30, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Li, Y.; Nishioka, K.; Daida, K.; Hayashida, A.; Ishiguro, Y.; Yamada, D.; Izawa, N.; Nishi, K.; Nishikawa, N.; et al. Genotype-phenotype correlation of Parkinson’s disease with PRKN variants. Neurobiol. Aging 2022, 114, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Huang, X.; Yoon, E.; Bandres-Ciga, S.; Blauwendraat, C.; Billingsley, K.J.; Cade, J.H.; Wu, B.P.; Williams, V.H.; Schindler, A.B.; et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease. Brain 2022, 145, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Denison, S.R.; Callahan, G.; Becker, N.A.; Phillips, L.A.; Smith, D.I. Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer 2003, 38, 40–52. [Google Scholar] [CrossRef]
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B.; et al. Distribution, type, and origin of Parkin mutations: Review and case studiesz. Mov. Disord. 2004, 19, 1146–1157. [Google Scholar] [CrossRef]
- Mefford, H.; Mitchell, E.; Hodge, J. 17q12 Recurrent Duplication. In GeneReviews(®); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chien, H.F.; Rohé, C.F.; Costa, M.D.L.; Breedveld, G.J.; Oostra, B.A.; Barbosa, E.R.; Bonifati, V. Early-onset Parkinson’s disease caused by a novel parkin mutation in a genetic isolate from north-eastern Brazil. Neurogenetics 2006, 7, 13–19. [Google Scholar] [CrossRef]
- Isaacs, D.; Claassen, D.; Bowman, A.B.; Hedera, P. Phenotypic Discordance in Siblings with Identical Compound Heterozygous PARK2 Mutations. Brain Sci. 2017, 7, 71. [Google Scholar] [CrossRef]
- Klein, C.; Pramstaller, P.P.; Kis, B.; Page, C.C.; Kann, M.; Leung, J.; Woodward, H.; Castellan, C.C.; Scherer, M.; Vieregge, P.; et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: Expanding the phenotype. Ann. Neurol. 2000, 48, 65–71. [Google Scholar] [CrossRef]
- Puschmann, A. Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Park. Relat. Disord. 2013, 19, 407–415. [Google Scholar] [CrossRef]
- Cukier, H.N.; Kim, H.; Griswold, A.J.; Codreanu, S.G.; Prince, L.M.; Sherrod, S.D.; McLean, J.A.; Dykxhoorn, D.M.; Ess, K.C.; Hedera, P.; et al. Genomic, transcriptomic, and metabolomic profiles of hiPSC-derived dopamine neurons from clinically discordant brothers with identical PRKN deletions. npj Park. Dis. 2022, 8, 84. [Google Scholar] [CrossRef]
- Periquet, M.; Lücking, C.B.; Vaughan, J.R.; Bonifati, V.; Dürr, A.; De Michele, G.; Horstink, M.W.; Farrer, M.; Illarioshkin, S.N.; Pollak, P.; et al. Origin of the mutations in the parkin gene in Europe: Exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am. J. Hum. Genet. 2001, 68, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Mutez, E.; Swiderski, M.; Devos, D.; Moreau, C.; Baille, G.; Degardin, A.; Ryckewaert, G.; Carriere, N.; Kreisler, A.; Simonin, C.; et al. Indication for molecular testing by multiplex ligation-dependent probe amplification in parkinsonism. Eur. J. Neurol. 2023, 30, 1667–1675. [Google Scholar] [CrossRef]
- Bravo, P.; Darvish, H.; Tafakhori, A.; Azcona, L.J.; Johari, A.H.; Jamali, F.; Paisán-Ruiz, C. Molecular characterization of PRKN structural variations identified through whole-genome sequencing. Mol. Genet. Genom. Med. 2018, 6, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Schaake, S.; Gabbert, C.; Lüth, T.; Cowley, S.A.; Fienemann, A.; Ullrich, K.K.; Klein, C.; Seibler, P. Optical genome mapping of structural variants in Parkinson’s disease-related induced pluripotent stem cells. BMC Genom. 2024, 25, 980. [Google Scholar] [CrossRef]
- Williams, E.S.; Barrett, M.J.; Dhamija, R.; Toran, L.; Chambers, C.; Mahadevan, M.S.; Golden, W.L. Phase determination using chromosomal microarray and fluorescence in situ hybridization in a patient with early onset Parkinson disease and two deletions in PRKN. Mol. Genet. Genom. Med. 2018, 6, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Seong, M.W.; Jeon, B.S.; Kim, S.Y.; Ko, H.S.; Kim, J.Y.; Park, S.S. Phase analysis identifies compound heterozygous deletions of the PARK2 gene in patients with early-onset Parkinson disease. Clin. Genet. 2012, 82, 77–82. [Google Scholar] [CrossRef]
- Daida, K.; Funayama, M.; Billingsley, K.J.; Malik, L.; Miano-Burkhardt, A.; Leonard, H.L.; Makarious, M.B.; Iwaki, H.; Ding, J.; Gibbs, J.R.; et al. Long-Read Sequencing Resolves a Complex Structural Variant in PRKN Parkinson’s Disease. Mov. Disord. 2023, 38, 2249–2257. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| SNP | Position | Mother | Proband | Father |
|---|---|---|---|---|
| rs6907465 | 162250726 | TT | AA | AT |
| rs9347543 | 162252602 | AA | GG | GA |
| rs9458393 | 162255535 | GG | TT | TG |
| rs6926642 | 162270981 | TT | CC | CT |
| rs9365329 | 162271943 | TT | GG | GT |
| rs9347547 | 162280309 | GG | AA | AG |
| rs7750426 | 162281372 | TT | GG | GT |
| rs2186803 | 162281492 | AA | CC | CA |
| rs2155486 | 162281533 | TT | AA | AT |
| rs6935164 | 162282924 | CC | TT | TC |
| rs9689946 | 162283949 | GG | TT | TG |
| rs9355958 | 162322332 | TT | CC | CC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choate, L.A.; Hoffman, F.; Newman, J.H.; Runke, C.; Webley, M.; Hoppman, N.L.; Thorland, E.C. Phase Determination and Demonstration of Parental Mosaicism of Intragenic PRKN Deletions Initially Identified by Chromosomal Microarray Analysis. Genes 2025, 16, 630. https://doi.org/10.3390/genes16060630
Choate LA, Hoffman F, Newman JH, Runke C, Webley M, Hoppman NL, Thorland EC. Phase Determination and Demonstration of Parental Mosaicism of Intragenic PRKN Deletions Initially Identified by Chromosomal Microarray Analysis. Genes. 2025; 16(6):630. https://doi.org/10.3390/genes16060630
Chicago/Turabian StyleChoate, Lauren A., Francis Hoffman, Jessica H. Newman, Cassandra Runke, Matthew Webley, Nicole L. Hoppman, and Erik C. Thorland. 2025. "Phase Determination and Demonstration of Parental Mosaicism of Intragenic PRKN Deletions Initially Identified by Chromosomal Microarray Analysis" Genes 16, no. 6: 630. https://doi.org/10.3390/genes16060630
APA StyleChoate, L. A., Hoffman, F., Newman, J. H., Runke, C., Webley, M., Hoppman, N. L., & Thorland, E. C. (2025). Phase Determination and Demonstration of Parental Mosaicism of Intragenic PRKN Deletions Initially Identified by Chromosomal Microarray Analysis. Genes, 16(6), 630. https://doi.org/10.3390/genes16060630