
Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Lithocarpus Species (Fagaceae) in South China

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Preservation of Materials
2.2. Chloroplast Genome Assembly and Comparative Analysis
2.2.1. DNA Extraction and Sequencing
2.2.2. Chloroplast Genome Assembly and Annotation
2.2.3. Chloroplast Genomes Comparison
2.2.4. IR Boundary Variation Analysis
2.2.5. Repetitive Sequence Analysis
2.2.6. Codon Usage Bias Analysis
2.2.7. Phylogenetic Tree Construction
3. Results
3.1. Chloroplast Genome Structure Characteristics
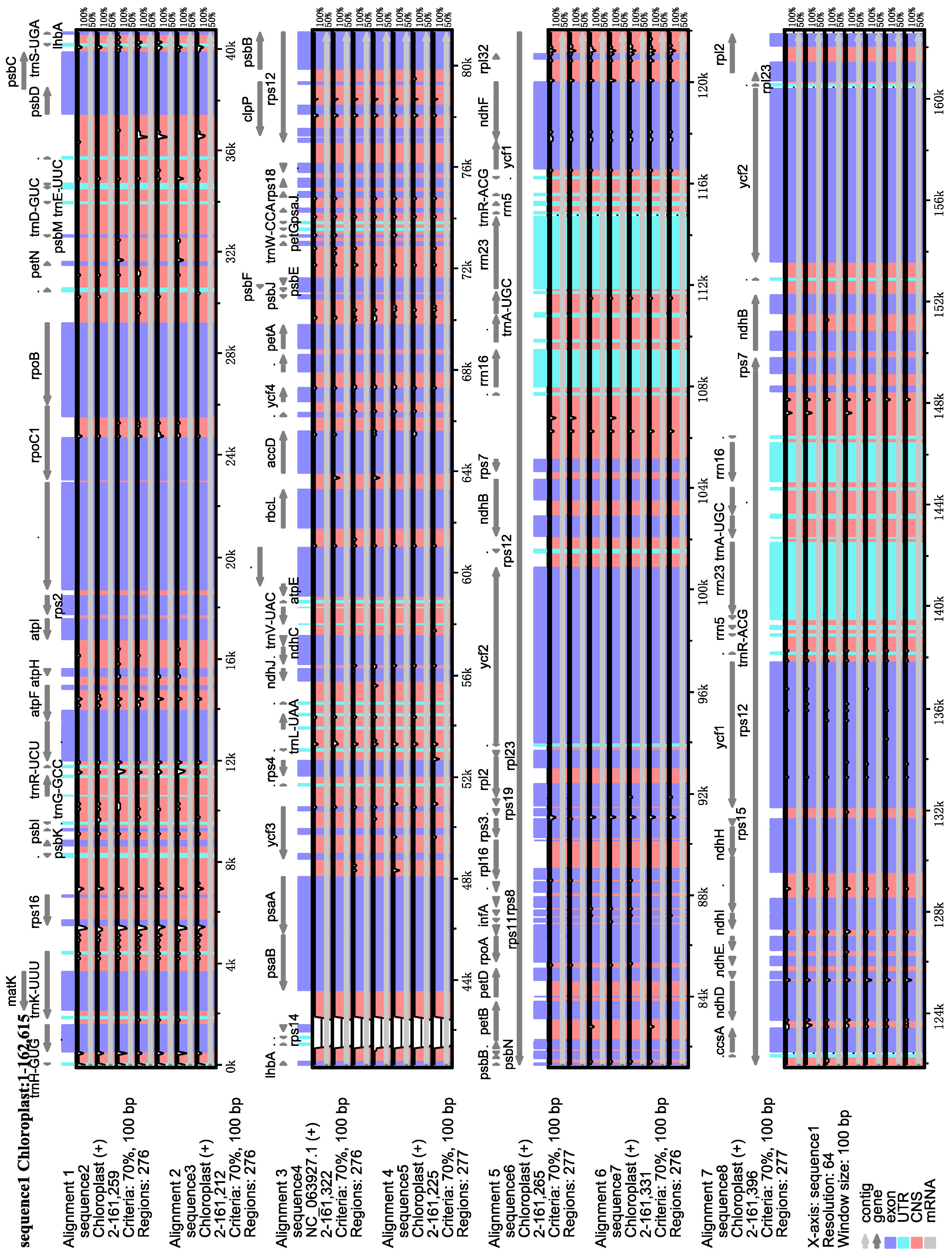
3.2. Comparative Analysis of Chloroplast Genomes
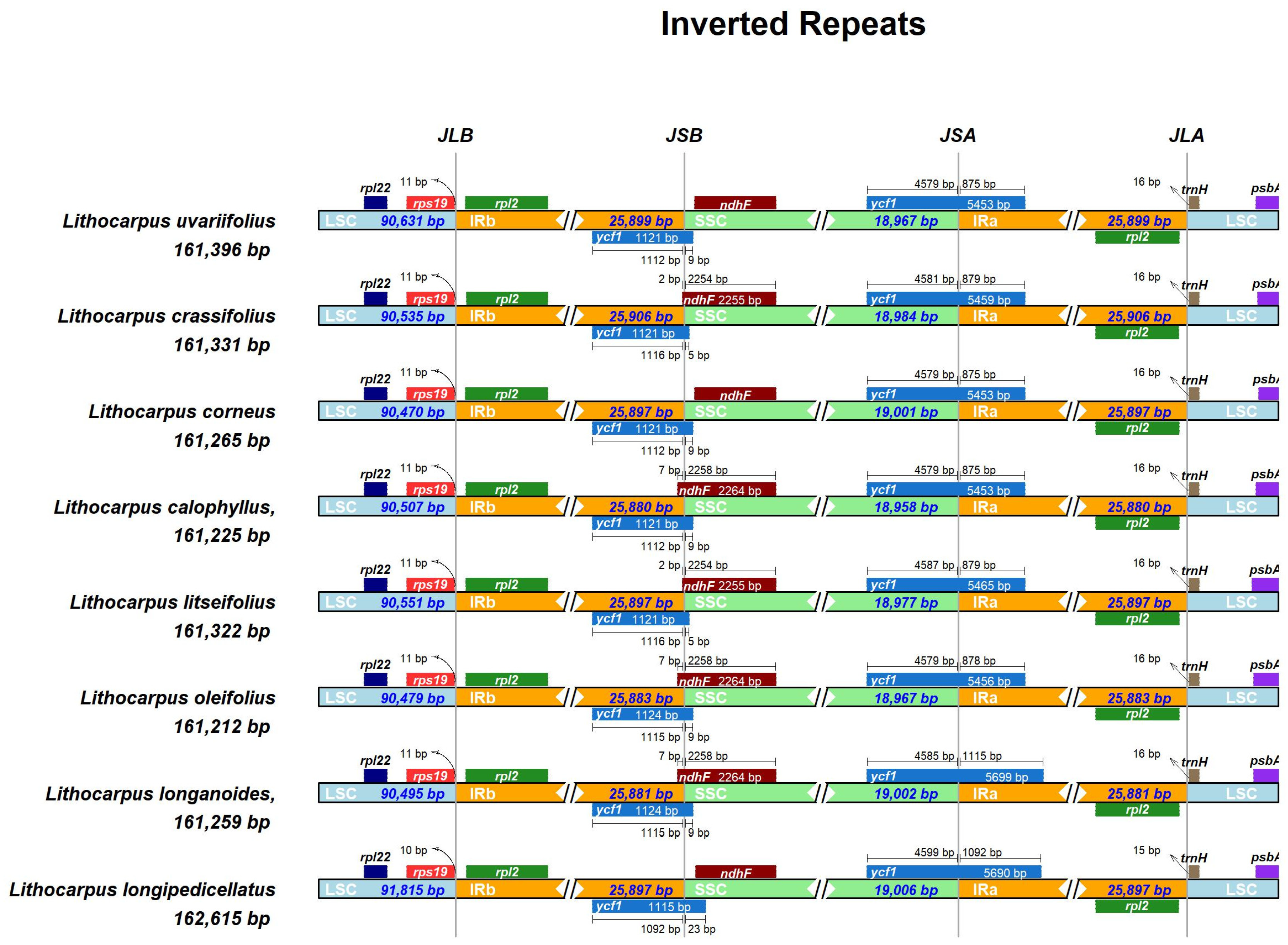
3.3. IR Region Contraction and Expansion (Analysis of IR Boundary Variation)
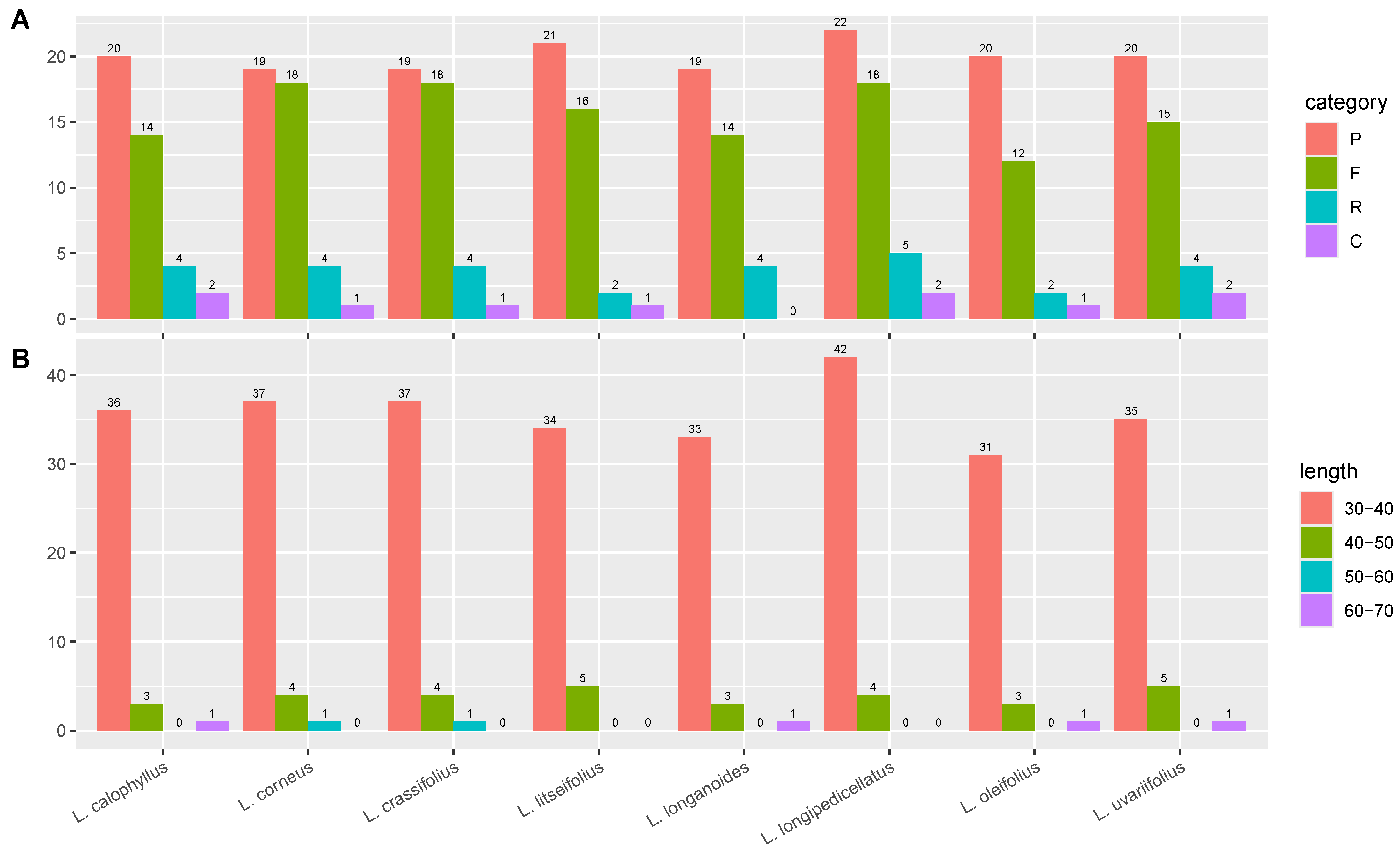
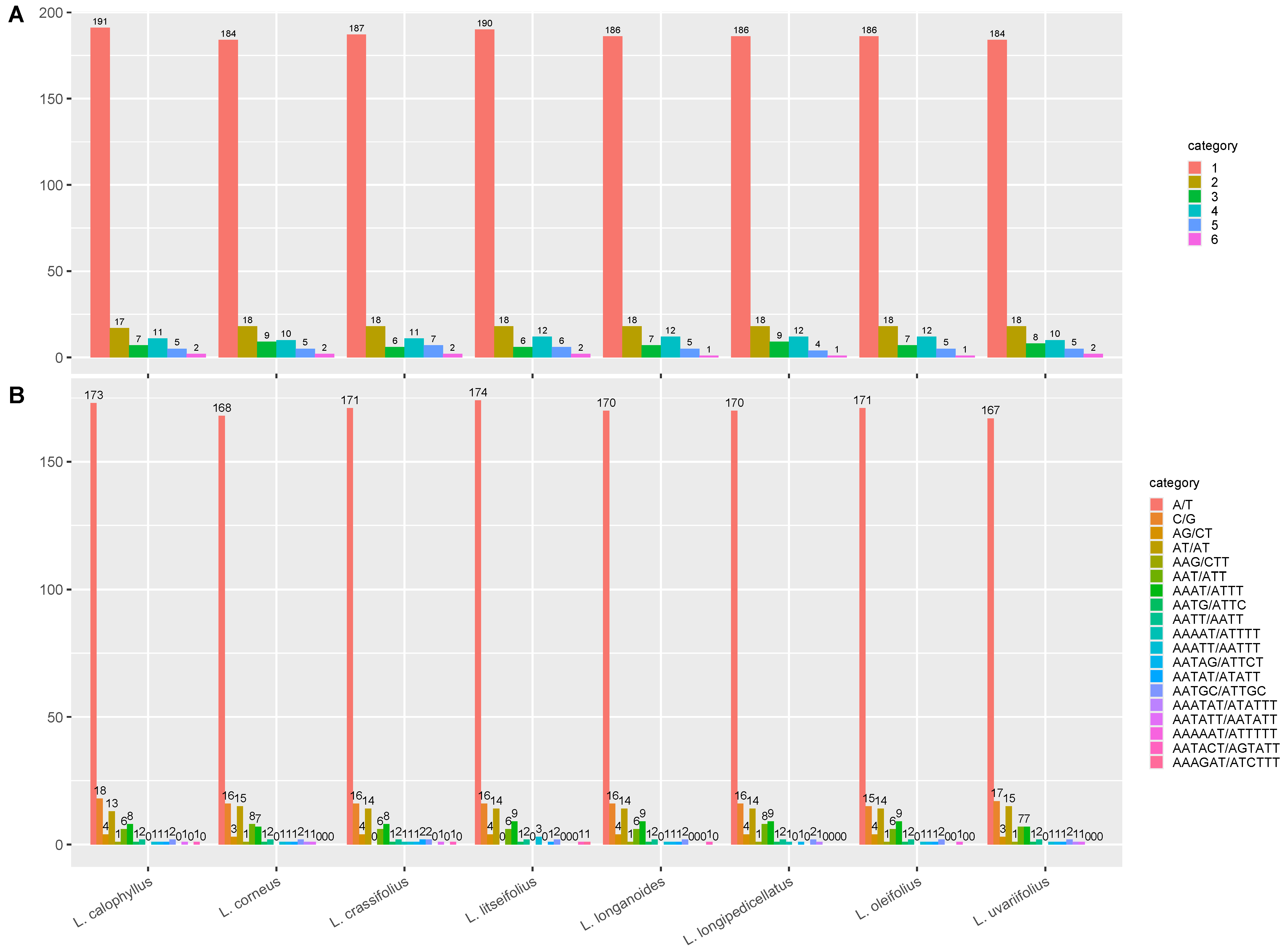
3.4. Repeat Analysis
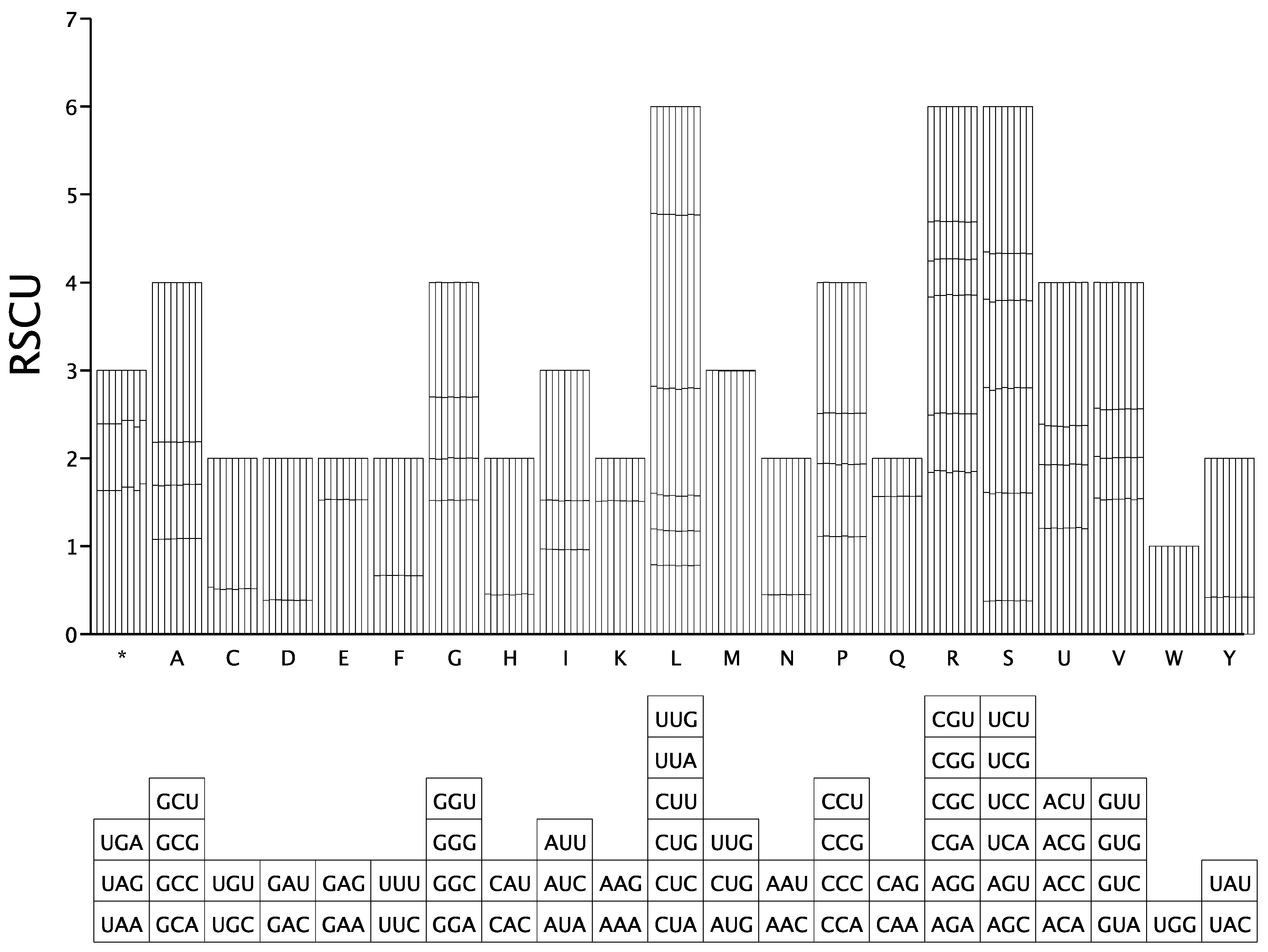
3.5. Code Usage Bias Analysis
3.6. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- POWO. Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. Available online: https://powo.science.kew.org/ (accessed on 9 April 2025).
- Webb, C.; Cannon, C.; Davies, S. Ecological organization, biogeography, and the phylogenetic structure of tropical forest tree communities. In Tropical Forest Community Ecology; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 79–97. [Google Scholar]
- Petit, R.J.; Carlson, J.; Curtu, A.L.; Loustau, M.L.; Plomion, C.; González-Rodríguez, A.; Sork, V.; Ducousso, A. Fagaceae trees as models to integrate ecology, evolution and genomics. New Phytol. 2013, 197, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.Y.; Li, J.X.; Xia, X.F.; Zhao, L.C. Cupules and fruits of Lithocarpus (Fagaceae) from the Miocene of Yunnan, southwestern China. Taxon 2015, 64, 795–808. [Google Scholar] [CrossRef]
- Cannon, C.H.; Manos, P.S. Phylogeography of the Southeast Asian stone oaks (Lithocarpus). J. Biogeogr. 2003, 30, 211–226. [Google Scholar] [CrossRef]
- Liu, X.Y.; Song, H.Z.; Jin, J.H. Diversity of Fagaceae on Hainan Island of South China During the Middle Eocene: Implications for Phytogeography and Paleoecology. Front. Ecol. Evol. 2020, 8, 255. [Google Scholar] [CrossRef]
- Wei, Z.; Baocheng, W.; Chunfeng, S.; Qixin, L. Resources and Exploitation of Lithocarpus(Fagaceae) in China. Chin. Wild Plant Resour. 2016, 35, 60–62. [Google Scholar]
- Guo, H.; Fu, M.X.; Zhao, Y.X.; Li, H.; Li, H.B.; Wu, D.T.; Gan, R.Y. The Chemical, Structural, and Biological Properties of Crude Polysaccharides from Sweet Tea (Lithocarpus litseifolius (Hance) Chun) Based on Different Extraction Technologies. Foods 2021, 10, 1779. [Google Scholar] [CrossRef]
- Hou, S.Z.; Chen, S.X.; Huang, S.; Jiang, D.X.; Zhou, C.J.; Chen, C.Q.; Liang, Y.M.; Lai, X.P. The hypoglycemic activity of Lithocarpus polystachyus Rehd. leaves in the experimental hyperglycemic rats. J. Ethnopharmacol. 2011, 138, 142–149. [Google Scholar] [CrossRef]
- Sun, Y.; Li, W.; Liu, Z. Preparative isolation, quantification and antioxidant activity of dihydrochalcones from Sweet Tea (Lithocarpus polystachyus Rehd.). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1002, 372–378. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, S.; Wu, C.; Jiang, X.; Deng, M. Plastome characterization and its phylogenetic implications on Lithocarpus (Fagaceae). BMC Plant Biol. 2024, 24, 1277. [Google Scholar] [CrossRef]
- Zhou, B.F.; Yuan, S.; Crowl, A.A.; Liang, Y.Y.; Shi, Y.; Chen, X.Y.; An, Q.Q.; Kang, M.; Manos, P.S.; Wang, B. Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nat. Commun. 2022, 13, 1320. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid Genome Comparative and Phylogenetic Analyses of the Key Genera in Fagaceae: Highlighting the Effect of Codon Composition Bias in Phylogenetic Inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Herliana, L.; Iryanto, S.; Fauzan, Y.; Robiansyah, I. Comparative analysis of Lithocarpus chloroplast genomes reveals candidate DNA barcoding loci. Proc. IOP Conf. Ser. Earth Environ. Sci. 2023, 1271, 012083. [Google Scholar] [CrossRef]
- Zhao, C.C.; Wang, Y.Y.; Chan, K.X.; Marchant, D.B.; Franks, P.J.; Randall, D.; Tee, E.E.; Chen, G.; Ramesh, S.; Phua, S.Y.; et al. Evolution of chloroplast retrograde signaling facilitates green plant adaptation to land. Proc. Natl. Acad. Sci. USA 2019, 116, 5015–5020. [Google Scholar] [CrossRef]
- Lu, R.; Hu, K.; Sun, X.; Chen, M. Low-coverage whole genome sequencing of diverse Dioscorea bulbifera accessions for plastome resource development, polymorphic nuclear SSR identification, and phylogenetic analyses. Front. Plant Sci. 2024, 15, 1373297. [Google Scholar] [CrossRef]
- Daniell, H.; Jin, S.; Zhu, X.G.; Gitzendanner, M.A.; Soltis, D.E.; Soltis, P.S. Green giant-a tiny chloroplast genome with mighty power to produce high-value proteins: History and phylogeny. Plant Biotechnol. J. 2021, 19, 430–447. [Google Scholar] [CrossRef]
- Lee, W.S.; Donrung, W.; Hung, B.M.; Muhamad Rusly, N.H.; Lee, S.Y.; Tanee, T. The complete plastid genome of Citrus hystrix DC. 1813 (Rutaceae) and its phylogenetic analysis. Mitochondrial DNA B Resour. 2025, 10, 94–98. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S.; Yi, T.S.; Li, D.Z.; Soltis, D.E. Plastome Phylogenetics: 30 Years of Inferences Into Plant Evolution—ScienceDirect. Adv. Bot. Res. 2018, 85, 293–313. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Qu, X.J.; Zhang, R.; Stull, G.W.; Yi, T.S. Plastid phylogenomic analyses of Fagales reveal signatures of conflict and ancient chloroplast capture. Mol. Phylogen. Evol. 2021, 163, 107232. [Google Scholar] [CrossRef]
- Chen, S.; Chen, R.; Zeng, X.; Chen, X.; Qin, X.; Zhang, Z.; Sun, Y. Genetic Diversity, Population Structure, and Conservation Units of Castanopsis sclerophylla (Fagaceae). Forests 2022, 13, 1239. [Google Scholar] [CrossRef]
- Liu, S.Y.; Yang, Y.Y.; Tian, Q.; Yang, Z.Y.; Li, S.F.; Valdes, P.J.; Farnsworth, A.; Kates, H.R.; Siniscalchi, C.M.; Guralnick, R.P.; et al. An integrative framework reveals widespread gene flow during the early radiation of oaks and relatives in Quercoideae (Fagaceae). J. Integr. Plant Biol. 2024, 67, 1119–1141. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Shi, L.C.; Chen, H.M.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731. [Google Scholar] [CrossRef]
- Jovanovic, N.; Mikheyev, A.S. Interactive web-based visualization and sharing of phylogenetic trees using phylogeny. IO. Nucleic Acids Res. 2019, 47, W266–W269. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Shelke, R.G.; Banerjee, R.P.; Joshi, B.; Singh, P.P.; Tiwari, G.J.; Adhikari, D.; Jena, S.N.; Barik, S.K. Chloroplast Genome of Lithocarpus dealbatus (Hook.f. & Thomson ex Miq.) Rehder Establishes Monophyletic Origin of the Species and Reveals Mutational Hotspots with Taxon Delimitation Potential. Life 2022, 12, 828. [Google Scholar] [CrossRef]
- Ma, C.X.; Yan, H.F.; Ge, X.J. The complete chloroplast genome of Lithocarpus hancei (Benth.) Rehd (Fagaceae) from Zhejiang, China. Mitochondrial DNA B Resour. 2021, 6, 2022–2023. [Google Scholar] [CrossRef]
- Liang, D.; Wang, H.; Zhang, J.; Zhao, Y.; Wu, F. Complete Chloroplast Genome Sequence of Fagus longipetiolata Seemen (Fagaceae): Genome Structure, Adaptive Evolution, and Phylogenetic Relationships. Life 2022, 12, 92. [Google Scholar] [CrossRef]
- Yu, R.; Huang, J.; Xu, Y.; Ding, Y.; Zang, R. Plant Functional Niches in Forests Across Four Climatic Zones: Exploring the Periodic Table of Niches Based on Plant Functional Traits. Front. Plant Sci. 2020, 11, 841. [Google Scholar] [CrossRef]
- Seol, M.-A.; Kim, S.S.; Lee, E.S.; Nam, K.-H.; Chun, S.-J.; Lee, J.-W. Yeast cadmium factor 1-enhanced oxidative stress tolerance in Arabidopsis thaliana. J. Plant Biotechnol. 2024, 51, 337–343. [Google Scholar] [CrossRef]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Li, Y.; Zheng, S.S.; Kozlowski, G.; Xu, J.; Song, Y.G. Complete Chloroplast Genomes of Four Oaks from the Section Cyclobalanopsis Improve the Phylogenetic Analysis and Understanding of Evolutionary Processes in the Genus Quercus. Genes. 2024, 15, 230. [Google Scholar] [CrossRef] [PubMed]
- Hubert, F.O.; Grimm, G.W.; Jousselin, E.; Berry, V.; Franc, A.; Kremer, A. Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst. Biodivers. 2014, 12, 405–423. [Google Scholar] [CrossRef]
- Oh, S.H.; Manos, P.S. Molecular phylogenetics and cupule evolution in Fagaceae as inferred from nuclear CRABS CLAW sequences. Taxon 2008, 57, 434–451. [Google Scholar] [CrossRef]
- Grimm, D.G.W. The oaks of western Eurasia: Traditional classifications and evidence from two nuclear markers. Taxon 2010, 59, 351–366. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Chloroplast Genome Size/bp | IR Length/bp | Overall GC Content % | Gene Number | tRNA Genes | Protein Coding Genes | GenBank Accession Number |
|---|---|---|---|---|---|---|---|
| L. longipedicellatus | 162,615 | 25,897 | 36.74 | 130 | 36 | 86 | PP234611.1 |
| L. longanoides | 161,259 | 25,881 | 36.75 | 130 | 36 | 86 | PV191269 |
| L. oleifolius | 161,212 | 25,883 | 36.74 | 130 | 36 | 86 | OR805597.1 |
| L. litseifolius | 161,322 | 25,897 | 36.73 | 130 | 36 | 86 | NC_063927.1 |
| L. calophyllus | 161,225 | 25,880 | 36.73 | 130 | 36 | 86 | PP234612.1 |
| L. corneus | 161,265 | 25,897 | 36.73 | 130 | 36 | 86 | PP234614.1 |
| L. crassifolius | 161,331 | 25,906 | 36.72 | 130 | 36 | 86 | PP234613.1 |
| L. uvariifolius | 161,396 | 25,899 | 36.76 | 130 | 36 | 86 | PP234615.1 |
| Category | Gene Group | Genes |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB *(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Subunits photochlorophyllide reductase | - | |
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16 *, rpl2 *(2), rpl20, rpl22, rpl23(2), rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | rps11, rps12 **(2), rps14, rps15, rps16 *, rps18, rps19, rps2, rps3, rps4, rps7(2), rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn4.5(2), rrn5(2) | |
| Transfer RNAs | trnA-UGC *(2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC *, trnG-UCC, trnH-GUG, trnI-CAU(2), trnI-GAU *(2), trnK-UUU *, trnL-CAA(2), trnL-UAA *, trnL-UAG, trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU(2), trnT-UGU, trnV-GAC(2), trnV-UAC *, trnW-CCA, trnY-GUA, trnfM-CAU | |
| Other genes | Maturase | matK |
| Protease | clpP ** | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | infA | |
| other | - | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | lhbA, ycf1(2), ycf2(2), ycf3 **, ycf4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.; Zhang, Z.; Lin, X.; Lu, L.; Fu, K.; He, M.; Lee, S.Y.; Yin, H.; Yu, J. Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Lithocarpus Species (Fagaceae) in South China. Genes 2025, 16, 616. https://doi.org/10.3390/genes16060616
Shi S, Zhang Z, Lin X, Lu L, Fu K, He M, Lee SY, Yin H, Yu J. Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Lithocarpus Species (Fagaceae) in South China. Genes. 2025; 16(6):616. https://doi.org/10.3390/genes16060616
Chicago/Turabian StyleShi, Shi, Ziyan Zhang, Xinhao Lin, Linjing Lu, Keyi Fu, Miaoxin He, Shiou Yih Lee, Hui Yin, and Jingwei Yu. 2025. "Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Lithocarpus Species (Fagaceae) in South China" Genes 16, no. 6: 616. https://doi.org/10.3390/genes16060616
APA StyleShi, S., Zhang, Z., Lin, X., Lu, L., Fu, K., He, M., Lee, S. Y., Yin, H., & Yu, J. (2025). Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Lithocarpus Species (Fagaceae) in South China. Genes, 16(6), 616. https://doi.org/10.3390/genes16060616