
The Complete Chloroplast Genome of Idesia polycarpa and Comparative Analysis of Related Species

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chloroplast Genome Sequencing, Assembly, and Annotation
2.3. Chloroplast Genome Characteristics
2.4. Repeat Sequence and Simple Repeat Sequence Analysis
2.5. Analysis of Codon Preference
2.6. Analysis of IR Boundary
2.7. Analysis of Selection Pressure
2.8. Comparative Analysis of CP Genomes
2.9. Phylogenetic Relationship Analysis
3. Results
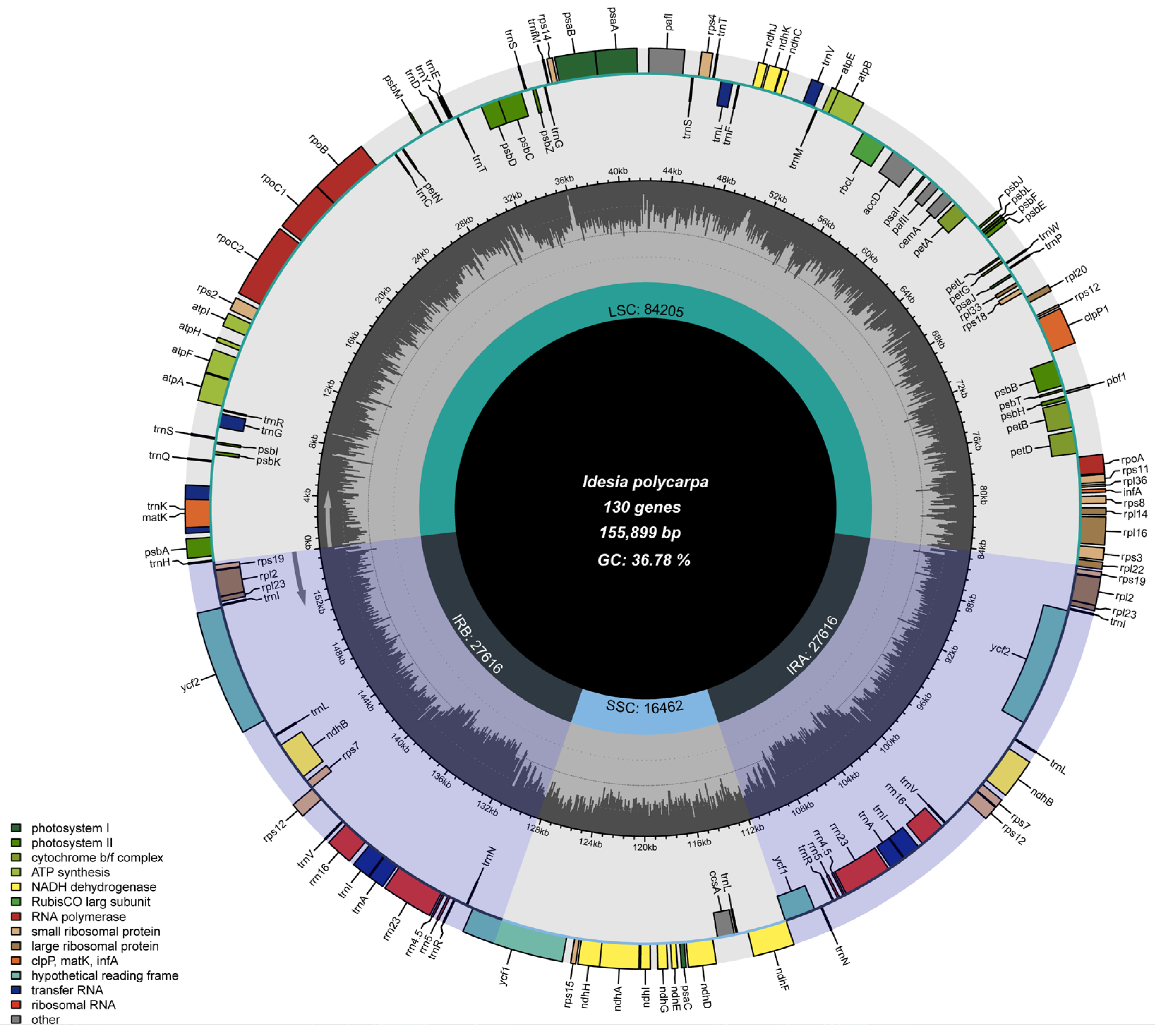
3.1. Chloroplast Genome Assembly and Genome Features
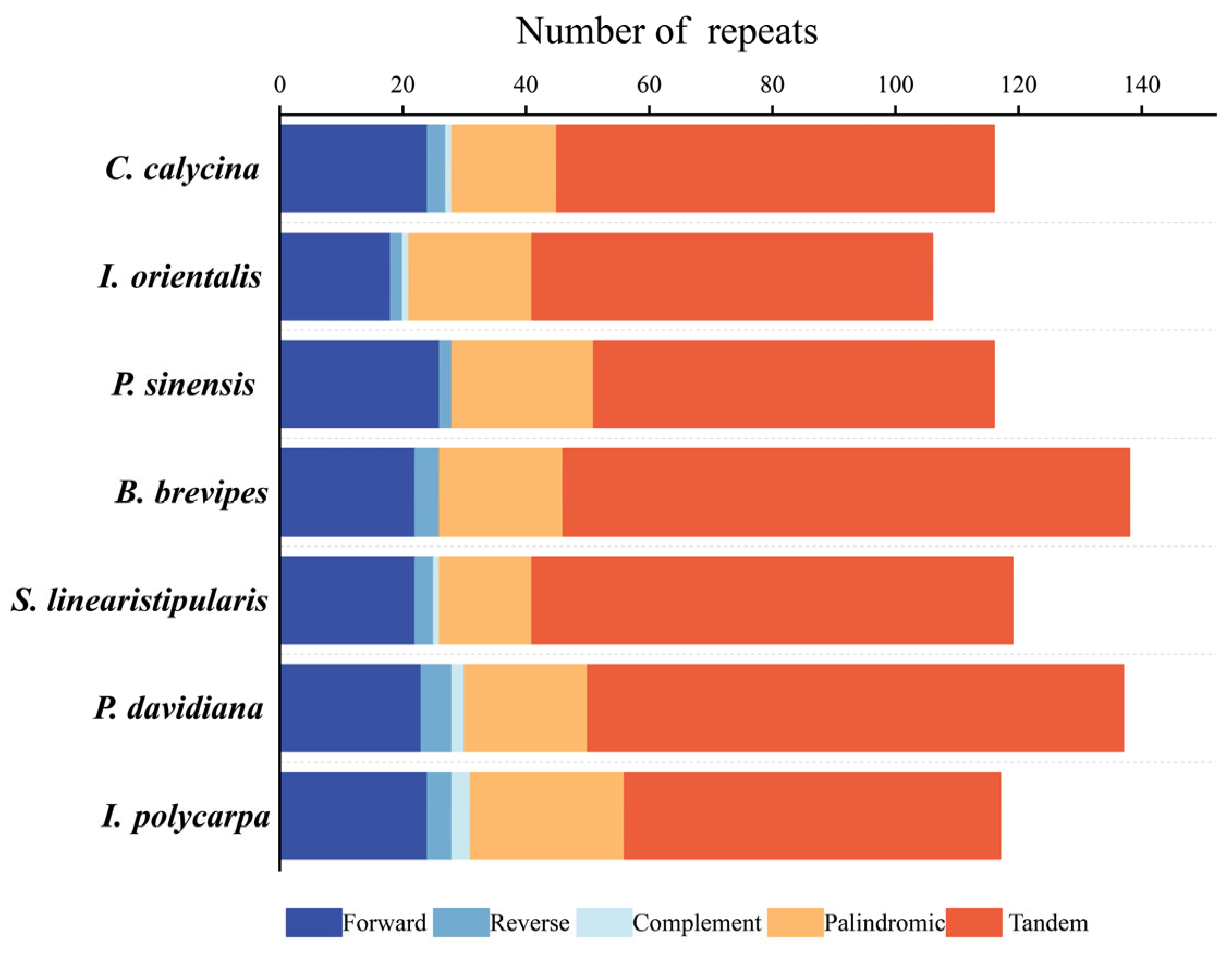
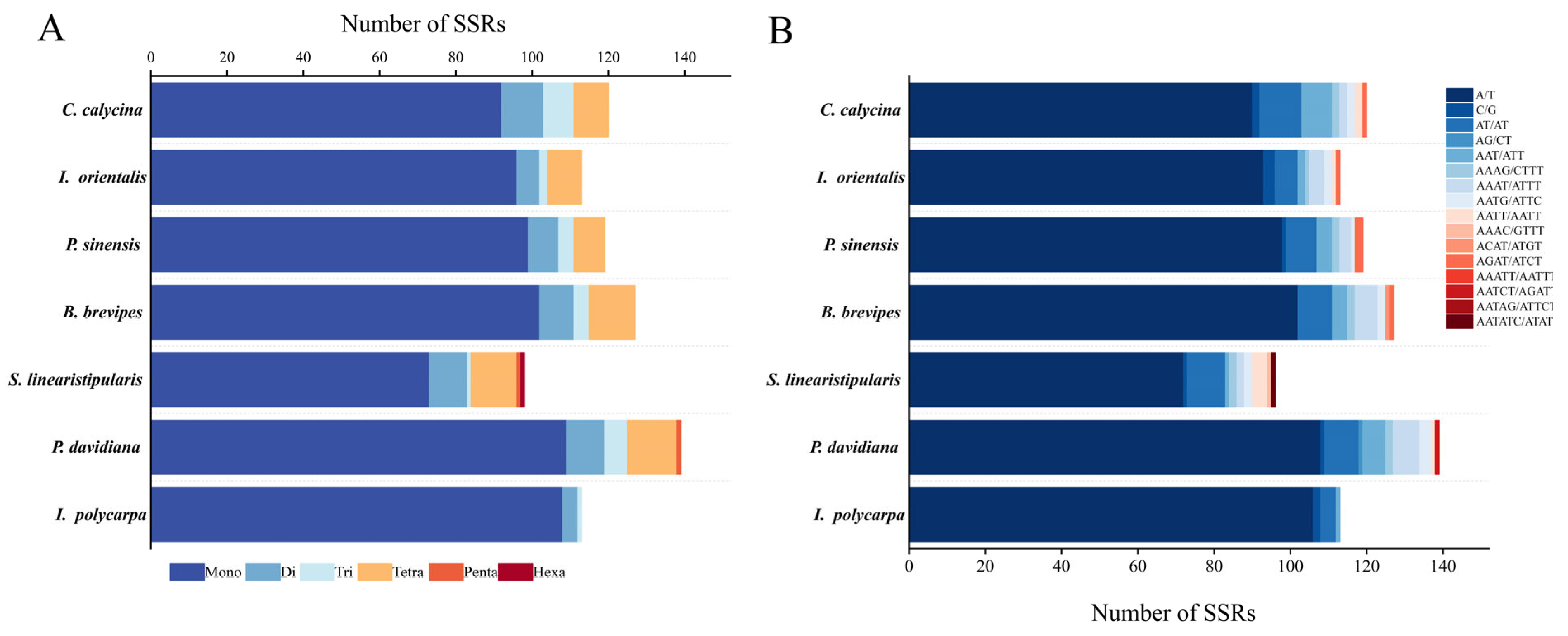
3.2. Repeat Sequences and SSR Analysis
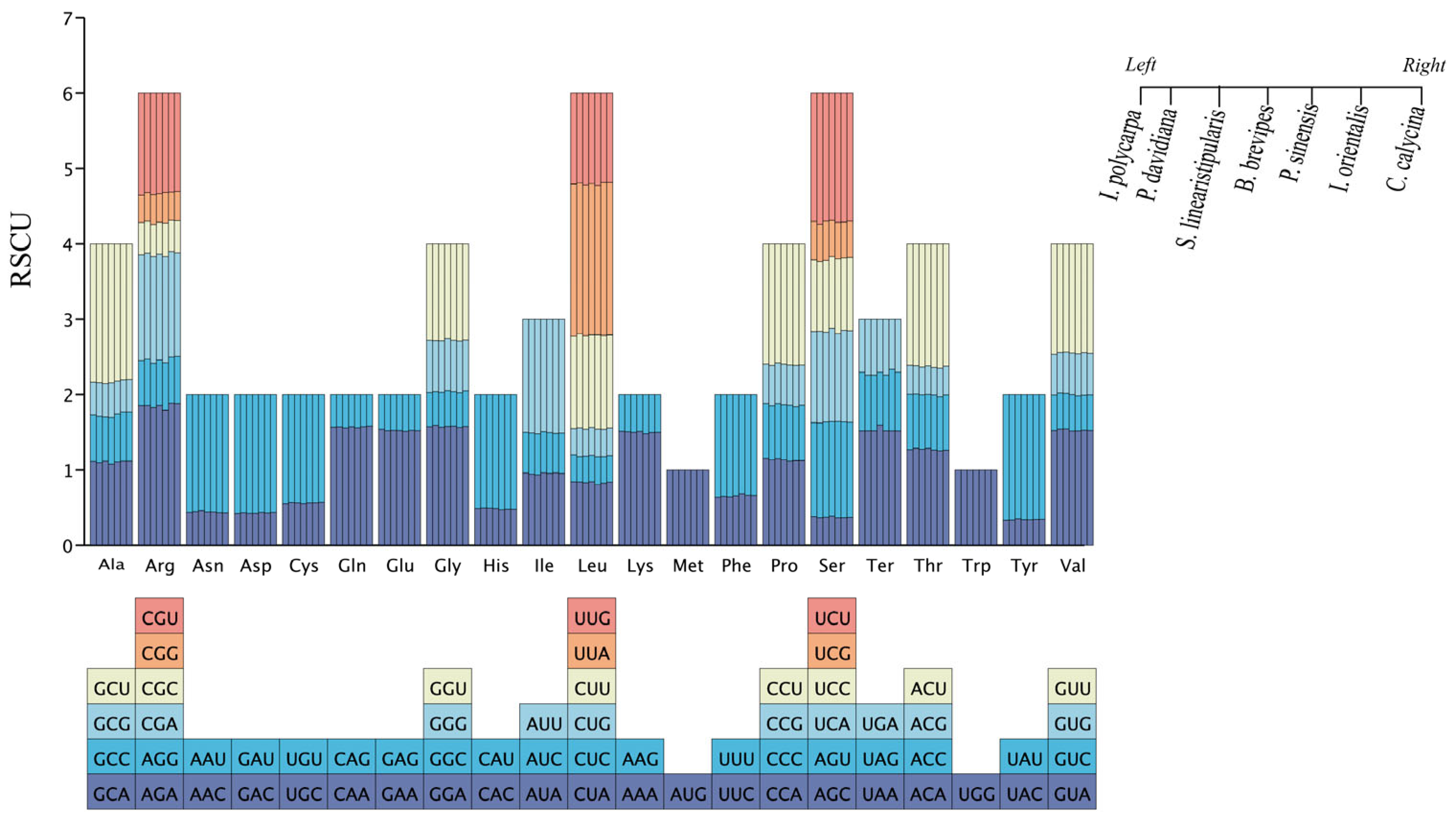
3.3. Codon Usage of Cp Genome
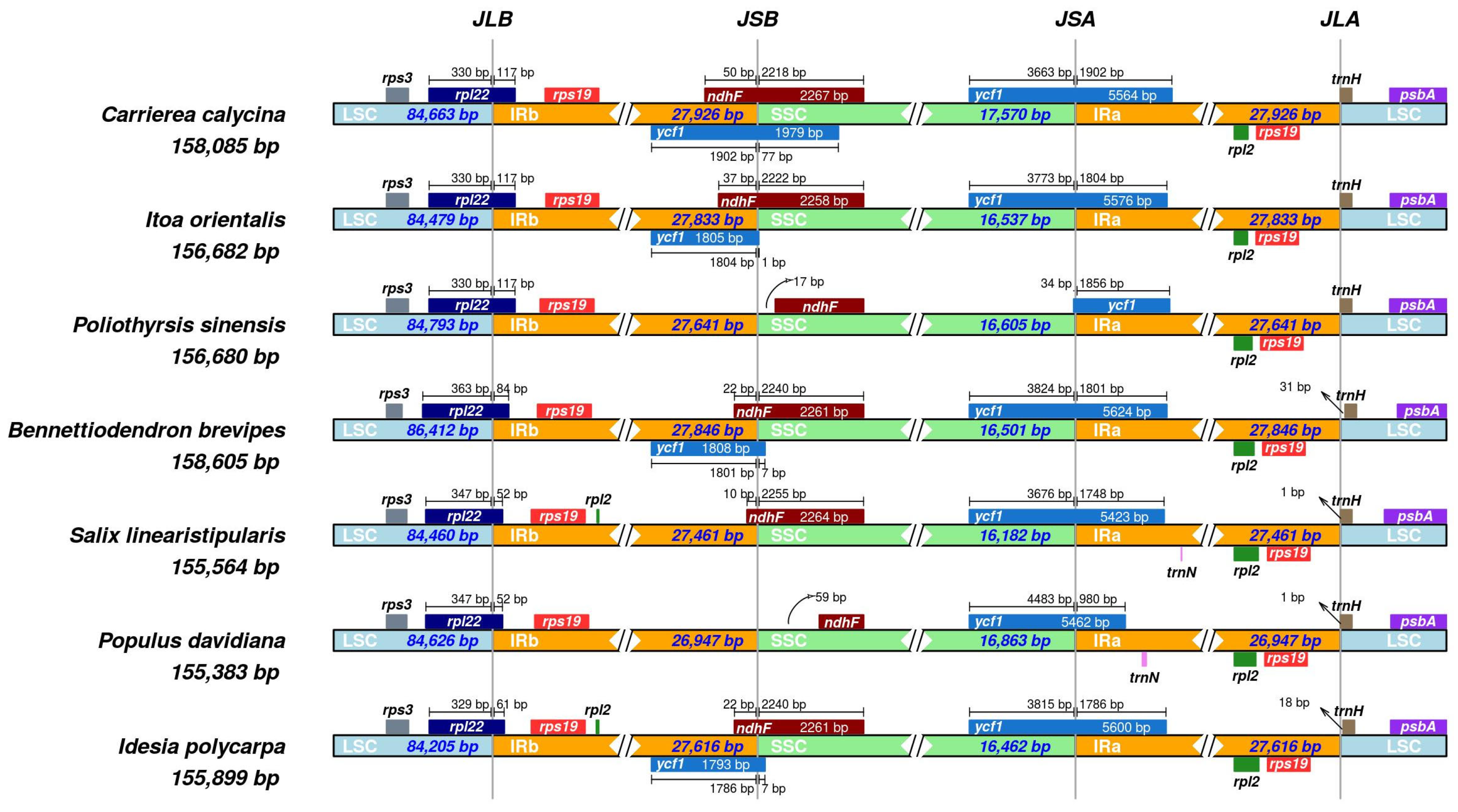
3.4. IR Boundary Analysis
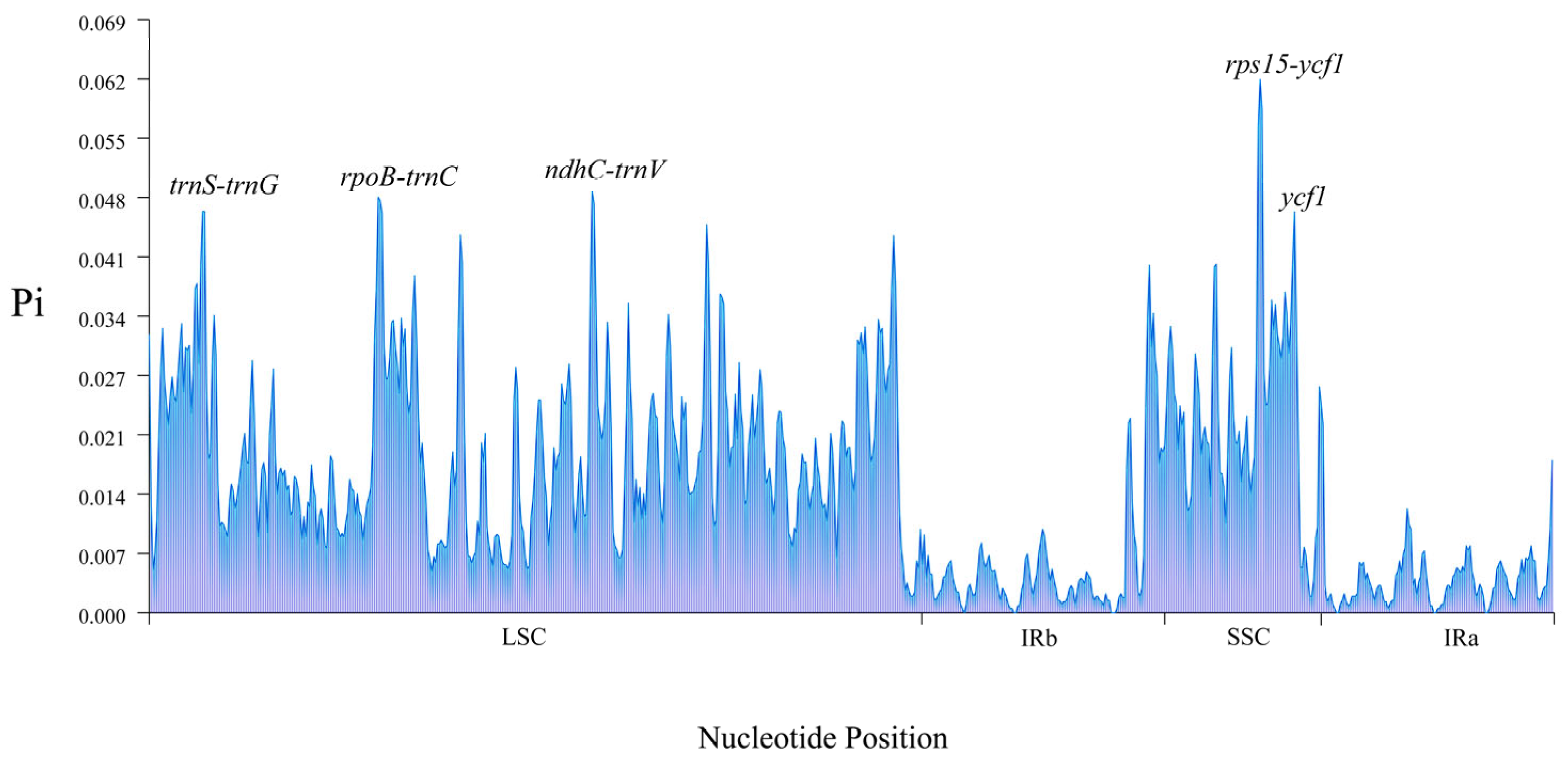
3.5. Comparative Analysis of Cp Genomes
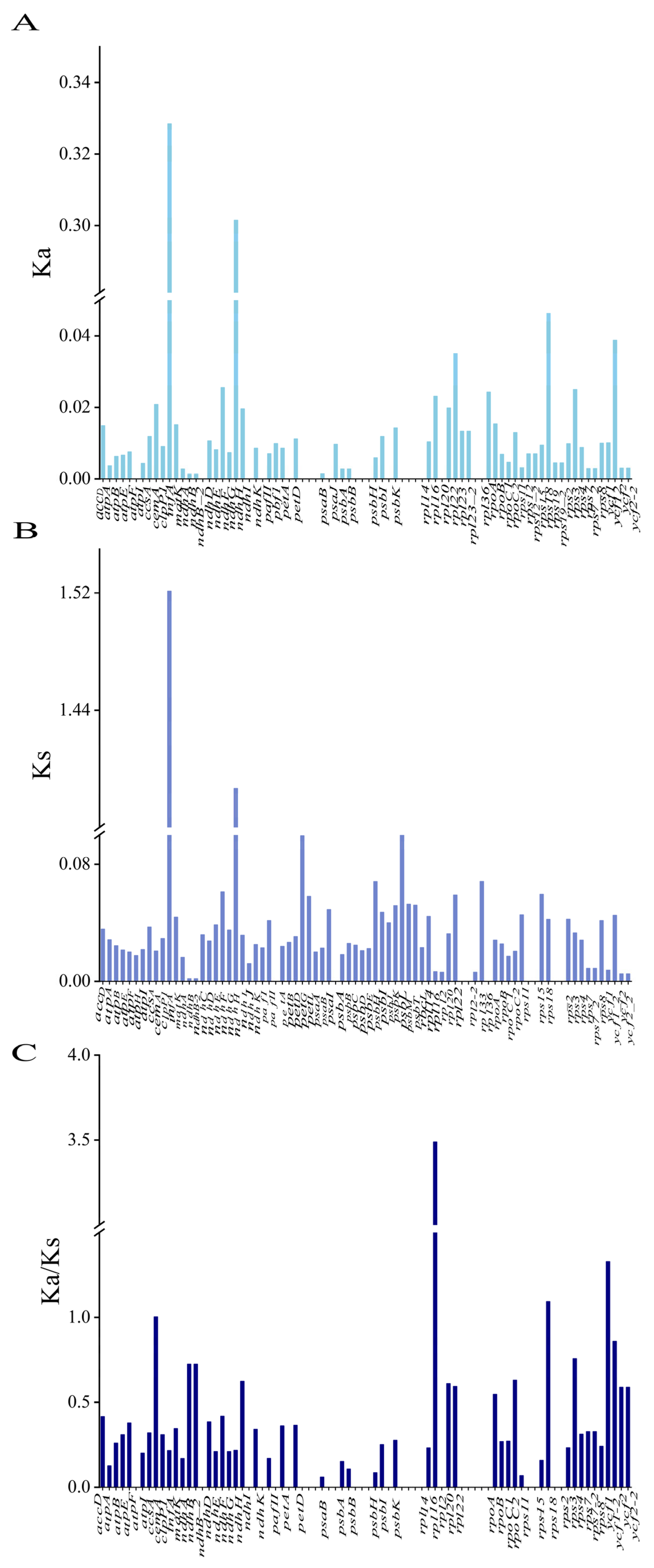
3.6. Selection Pressure Analysis
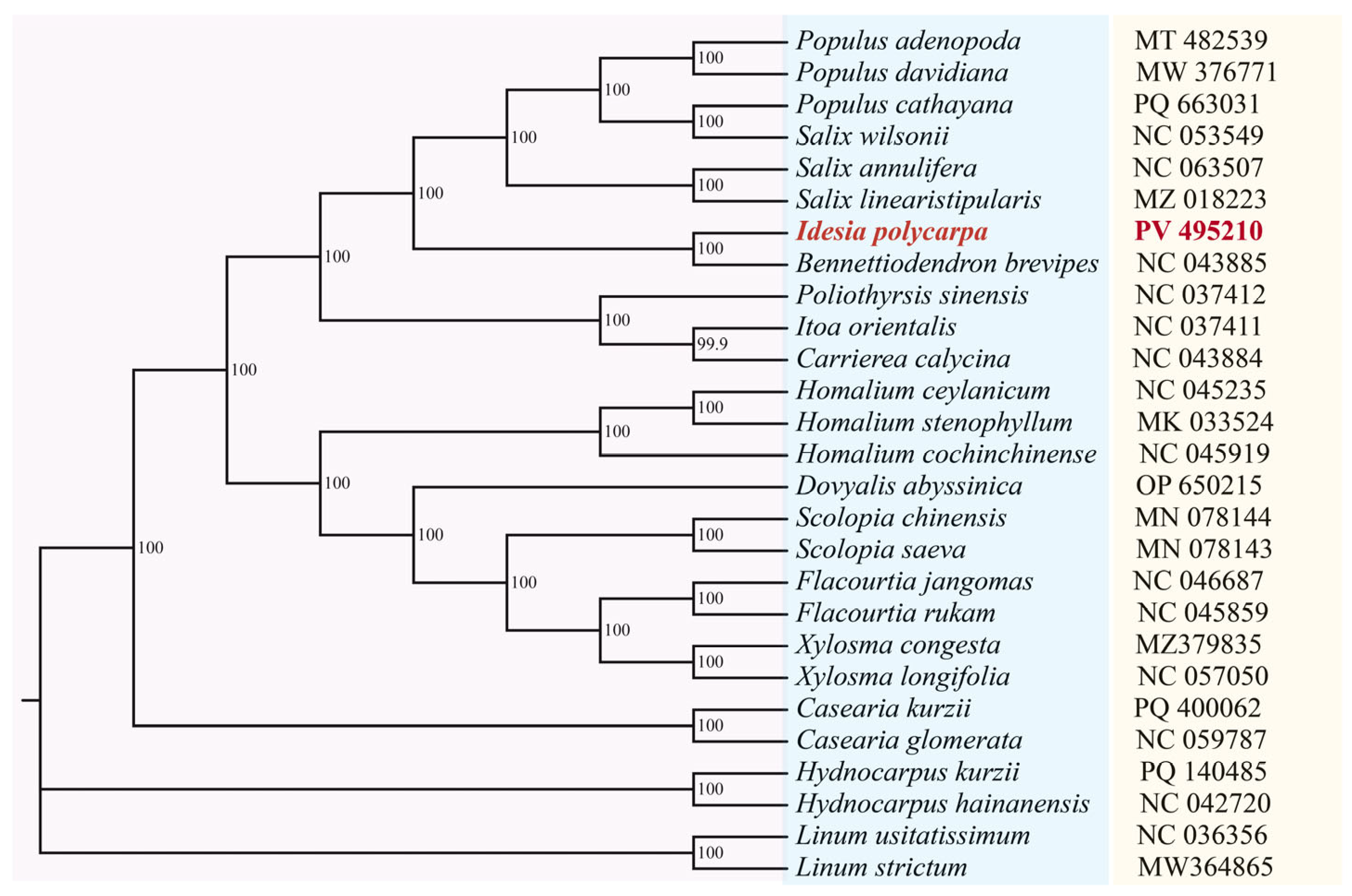
3.7. Phylogenetic Analysis
4. Discussion
4.1. Structural Features of the Cp Genome in Idesia polycarpa
4.2. Phylogenetic Relationship Between Idesia polycarpa and Related Species
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, L.-Y.; Mao, J.-M.; An, X.-F.; Yang, B.; Wang, Z.-R.; Kan, J.-Q. Dynamic analysis and evaluation of chemical constituents and fatty acids of Idesia polycarpa var. vestita Diels fruit. Food Sci. 2022, 43, 198–207. [Google Scholar] [CrossRef]
- Liu, X.-M. Study on Propagation and Tissue Culture Techniques of Idesia polycarpa Maxim. Master’s Thesis, Northwest University of Agriculture and Forestry Science and Technology, Yangling, China, 2009. [Google Scholar]
- Yang, E.-R.; Liu, X.-M.; Xie, Q. Establishment of plantlet regeneration system of Idesia polycarpa in vitro. J. Northwest For. Univ. 2013, 28, 95–98. [Google Scholar] [CrossRef]
- Jiang, X.-B.; Gong, B.-C.; Li, D.-W.; Wu, K.-Y.; Zhao, X.-M. Variation analysis of phenotypic traitsin natural population of Idesia polycarpa. For. Res. 2013, 26, 113–117. [Google Scholar] [CrossRef]
- Jia, R.-R.; Tang, X.-S.; Dong, N.; Tang, L.; Chen, F. Influence of different mature degrees on seeds germination and quality of fruit oil of Idesia polycarpa Maxim. var. vestita diels. Seed 2014, 33, 38–41. [Google Scholar] [CrossRef]
- Fan, R.-S.; Li, L.; Cai, G.; Ye, J.; Liu, M.-H.; Wang, S.-H.; Li, Z.-Q. Molecular cloning and function analysis of FAD2 gene in Idesia polycarpa. Phytochemistry 2019, 168, 112114. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Li, Z.-J.; Ni, J.-W.; Geng, Y.-H.; Xu, X.-Q. Diversity analysis on characters of spikes and fruits of Idesia polycarpa. J. Plant Resour. Environ. 2021, 33, 35–44. [Google Scholar] [CrossRef]
- Cai, Q.-F.; Zhang, M.-M.; Xu, Z.-H.; Wang, S.; Xu, D.-H.; Liu, Z. Genetic diversity analysis of germplasm resources of Idesia polycarpa Maxim. J. Henan Agric. Univ. 2023, 57, 47–55. [Google Scholar] [CrossRef]
- Ji, Y.-F.; Song, S.-Q.; Liu, J.; Tian, X.-R. Study on characteristics of dormancy and germination in Idesia polycarpa seeds. Chin. Wild Plant Resour. 2023, 42, 23–28. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Liu, C.-Y.; Hu, J.-S.; Wu, K.-Y.; Gong, B.-C.; Xu, Y. A flowering morphological investigation, fruit fatty acids, and mineral elements dynamic changes of Idesia polycarpa Maxim. Plants 2024, 13, 2663. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, S.-S.; Rana, S.; Wang, Y.-M.; Liu, Z.; Cai, Q.-F.; Geng, X.-D.; Yuan, Q.-P.; Yang, Y.; Miao, C.; et al. Analysis of leaf and soil nutrients, microorganisms and metabolome in the growth period of Idesia polycarpa Maxim. Microorganisms 2024, 12, 746. [Google Scholar] [CrossRef]
- Ping, H.-R.; Ge, Y.-H.; Liu, W.-X.; Yang, J.-X.; Zhong, Z.-X.; Wang, J.-H. Effects of Different drying methods on volatile flavor compounds in Idesia Polycarpa Maxim fruit and oil. Molecules 2025, 30, 811. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.-T.; Xu, J.-Q. Quality analysis of Idesia polycarpa fruit oil samples from cultivars with different phenotypes. Food Chem. X 2025, 25, 102127. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Wang, J.; Kan, S.-L.; Liao, X.-Z.; Zhou, J.-W.; Tembrock, L.R.; Daniell, H.; Jin, S.-X.; Wu, Z.-Q. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Zhou, J.W.; He, W.-C.; Wang, J.; Liao, X.-Z.; Xiang, K.-L.; Ma, M.-C.; Liu, Z.; Li, Y.-Y.; Tembrock, L.R.; Wu, Z.-Q.; et al. The pan-plastome of tartary buckwheat (Fagopyrum tataricum): Key insights into genetic diversity and the history of lineage divergence. BMC Plant Biol. 2023, 23, 212. [Google Scholar] [CrossRef]
- Jia, M.-L.; Wang, J.; Cao, D.-M.; Jiang, C.-R.; Li, W.; Tembrock, L.-R.; Xing, G.-M.; Li, S.; Wu, Z.-Q. The pan-plastome of Hemerocallis citrina reveals new insights into the genetic diversity and cultivation history of an economically important food plant. BMC Plant Biol. 2024, 24, 44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, J.-H.; Wang, J.; Yan, X.-L.; Li, Y.; Soe, T.; Tembrock, L.R.; Xing, G.-M.; Li, S.; Wu, Z.-Q.; et al. The pan-plastome of Prunus mume: Insights into Prunus diversity, phylogeny, and domestication history. Front. Plant Sci. 2024, 15, 1404071. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Liu, H.-B.; Li, B.; Zhao, H.; Li, X.-L.; Chen, J.-T.; Wang, L.; Zheng, Q.-B.; He, Y.-Q.; Zhang, J.-S.; et al. The Idesia polycarpa genome provides insights into its evolution and oil biosynthesis. Cell Rep. 2024, 43, 113909. [Google Scholar] [CrossRef]
- Yang, W.-L.; Wang, W.-W.; Zhang, L.; Chen, Z.-Y.; Guo, X.-Y.; Ma, T. Characterization of the complete chloroplast genome of Idesia polycarpa. Conser. Gen. Res. 2016, 8, 271–273. [Google Scholar] [CrossRef]
- Kikuchi, T.; Miura, O. Differentiation in vegetation related to micro-scale landforms with special reference to the lower side slope. Ecol. Rev. 1991, 22, 61–70. [Google Scholar]
- Shimada, K. Topographical distribution of five pioneer tree species and significance of their tree forms in natural forests on Mt. Takao, central Japan. Jpn J. Ecol. 1994, 44, 293–304. [Google Scholar] [CrossRef]
- Huang, X.-M.; Qiu, Z.-Q.; Wang, J.-J.; Wu, Y. Research progress on oil from Idesia polycarpa. Cereals Oils. 2017, 30, 11–13. [Google Scholar] [CrossRef]
- Wang, Z.-X. Handbook of Chinese Resource Plants, 1st ed.; Science Press: Beijing, China, 1989; pp. 233–234. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Son, P.P.; Andrey, D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect trna genes and tmrna genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Burland, T.G. DNASTAR’s lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellargenomedraw (ogdraw): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z. Kaks_calculator 3.0: Calculating selective pressure on coding and noncoding sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Carlos Sanchez-DelBarrio, J.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Sun, C.; Li, H.-L.; Li, Z.-X.; Guo, Y.; Fu, X.-Q.; Liao, Q.-H.; Zhang, W.-L.; Liu, Y.-Q. Chloroplast genome comparison and phylogenetic analysis of the commercial variety Actinidia chinensis ‘Hongyang’. Genes 2023, 14, 2136. [Google Scholar] [CrossRef]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef]
- Guo, Q.; Cao, S.; Li, D.; Li, X.-Y.; Zhang, J.-T.; Zhang, Y.-S.; Zhang, Z.-J.; Sun, Y.-H.; Cui, L.; Fan, Y.-M.; et al. Genetic diversity and population structure of Robinia pseudoacacia from six improved variety bases in China as revealed by simple sequence repeat markers. J. For. Res. 2022, 33, 611–621. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.-P.; Guo, X.; Liu, Q.-H.; Wang, K.-L. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 2019, 14, e0216645. [Google Scholar] [CrossRef]
- Cui, Y.-X.; Nie, L.-P.; Sun, W.; Xu, Z.-C.; Wang, Y.; Yu, J.; Song, J.-Y.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Androsiuk, P.; Jastrzebski, J.P.; Paukszto, L.; Makowczenko, K.; Okorski, A.; Pszczolkowska, A.; Chwedorzewska, K.J.; Gorecki, R.; Gielwanowska, I. Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species. Sci. Rep. 2020, 10, 11522. [Google Scholar] [CrossRef]
- Zhao, J.-T.; Xu, Y.; Xi, L.-J.; Yang, J.-W.; Chen, H.-W.; Zhang, J. Characterization of the chloroplast genome sequence of Acer miaotaiense: Comparative and phylogenetic analyses. Molecules 2018, 23, 1740. [Google Scholar] [CrossRef]
- Li, W.; Wu, C.; Luo, C.-C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Nam, G.H.; Park, S. Extensive reorganization of the chloroplast genome of Corydalis platycarpa: A comparative analysis of their organization and evolution with other Corydalis plastomes. Front. Plant Sci. 2022, 13, 1043740. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Barton, N.H. Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes. Science 2003, 300, 321–324. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Saina, J.K.; Gichira, A.W.; Li, Z.-Z.; Hu, G.-W.; Wang, Q.-F.; Liao, K. The complete chloroplast genome sequence of Dodonaea viscosa: Comparative and phylogenetic analyses. Genetica 2017, 146, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Han, L.; Chen, C.; Wang, Z.-Z. The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS ONE 2018, 13, e0192956. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z.-S.; Wang, D.-S.; Zhang, J.-G. Phylogeny of Populus-Salix (Salicaceae) and their relative genera using molecular datasets. Biochem. Syst. Ecol. 2016, 68, 210–215. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Group | Gene Name | Number |
|---|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ | 14 | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB *(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK, | 12 | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Subunits photochlorophyllide reductase | —— | ||
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16 *, rpl2 *(2), rpl20, rpl22, rpl23(2), rpl33, rpl36 | 10 |
| Proteins of small ribosomal subunit | rps11, rps12 **(2), rps14, rps15, rps18, rps19(2), rps2, rps3, rps4, rps7(2), rps8 | 14 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | 4 | |
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn4.5(2), rrn5(2) | 8 | |
| Transfer RNAs | trnA-UGC *(2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU(2), trnI-GAU *(2), trnK-UUU *, trnL-CAA(2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC *, trnW-CCA, trnY-GUA, trnfM-CAU | 37 | |
| Other genes | Maturase | matK | 1 |
| Protease | clpP1 ** | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Acetyl-CoA carboxylase | accD | 1 | |
| c-type cytochrome synthesis gene | ccsA | 1 | |
| Translation initiation factor | infA | 1 | |
| other | pafI **, pafII, pbf1 | 3 | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | ycf1(2), ycf2(2) | 4 |
| Gene | Location | Exon1 | Intron1 | Exon2 | Intron2 | Exon3 |
|---|---|---|---|---|---|---|
| trnK(UUU) | LSC | 37 | 2544 | 35 | ||
| trnG(UCC) | LSC | 23 | 708 | 48 | ||
| atpF | LSC | 145 | 734 | 410 | ||
| rpoC1 | LSC | 432 | 776 | 1617 | ||
| pafI | LSC | 124 | 722 | 230 | 712 | 153 |
| trnL(UAA) | LSC | 35 | 581 | 50 | ||
| trnV(UAC) | LSC | 39 | 594 | 35 | ||
| rps12 | LSC + IRa | 114 | 536 | 232 | 26 | |
| rps12-2 | LSC + IRb | 114 | 536 | 232 | 26 | |
| clpP1 | LSC | 71 | 762 | 292 | 640 | 228 |
| petB | LSC | 6 | 814 | 642 | ||
| petD | LSC | 8 | 490 | |||
| rpl16 | LSC | 9 | 1102 | 399 | ||
| rpl2 | IRa | 391 | 668 | 434 | ||
| ndhB | IRa | 777 | 682 | 756 | ||
| trnI(GAU) | IRa | 37 | 948 | 35 | ||
| trnA(UGC) | IRa | 38 | 809 | 35 | ||
| ndhA | SSC | 553 | 1033 | 545 | ||
| trnA(UGC)-2 | IRb | 38 | 809 | 35 | ||
| trnI(GAU)-2 | IRb | 37 | 948 | 35 | ||
| ndhB-2 | IRb | 777 | 682 | 756 | ||
| rpl2-2 | IRb | 391 | 668 | 434 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, X.; Luo, J.; Guo, Y.; Feng, D.; Deng, Y.; Kuang, M.; Zhou, H.; Liu, X.; Sun, C. The Complete Chloroplast Genome of Idesia polycarpa and Comparative Analysis of Related Species. Genes 2025, 16, 611. https://doi.org/10.3390/genes16050611
Fu X, Luo J, Guo Y, Feng D, Deng Y, Kuang M, Zhou H, Liu X, Sun C. The Complete Chloroplast Genome of Idesia polycarpa and Comparative Analysis of Related Species. Genes. 2025; 16(5):611. https://doi.org/10.3390/genes16050611
Chicago/Turabian StyleFu, Xueqian, Jie Luo, Yuan Guo, Dalan Feng, Yifei Deng, Mi Kuang, Houlin Zhou, Xia Liu, and Chong Sun. 2025. "The Complete Chloroplast Genome of Idesia polycarpa and Comparative Analysis of Related Species" Genes 16, no. 5: 611. https://doi.org/10.3390/genes16050611
APA StyleFu, X., Luo, J., Guo, Y., Feng, D., Deng, Y., Kuang, M., Zhou, H., Liu, X., & Sun, C. (2025). The Complete Chloroplast Genome of Idesia polycarpa and Comparative Analysis of Related Species. Genes, 16(5), 611. https://doi.org/10.3390/genes16050611