

Deciphering the First Mitochondrial Genome of the Liolophura Pilsbry, 1893 Genus: An Extensive Phylogenetic Study Within the Chitonidae Family

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Specimen Collection and DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Genome Assembly and Annotation
2.5. Sequence and Phylogenetic Analysis
3. Results
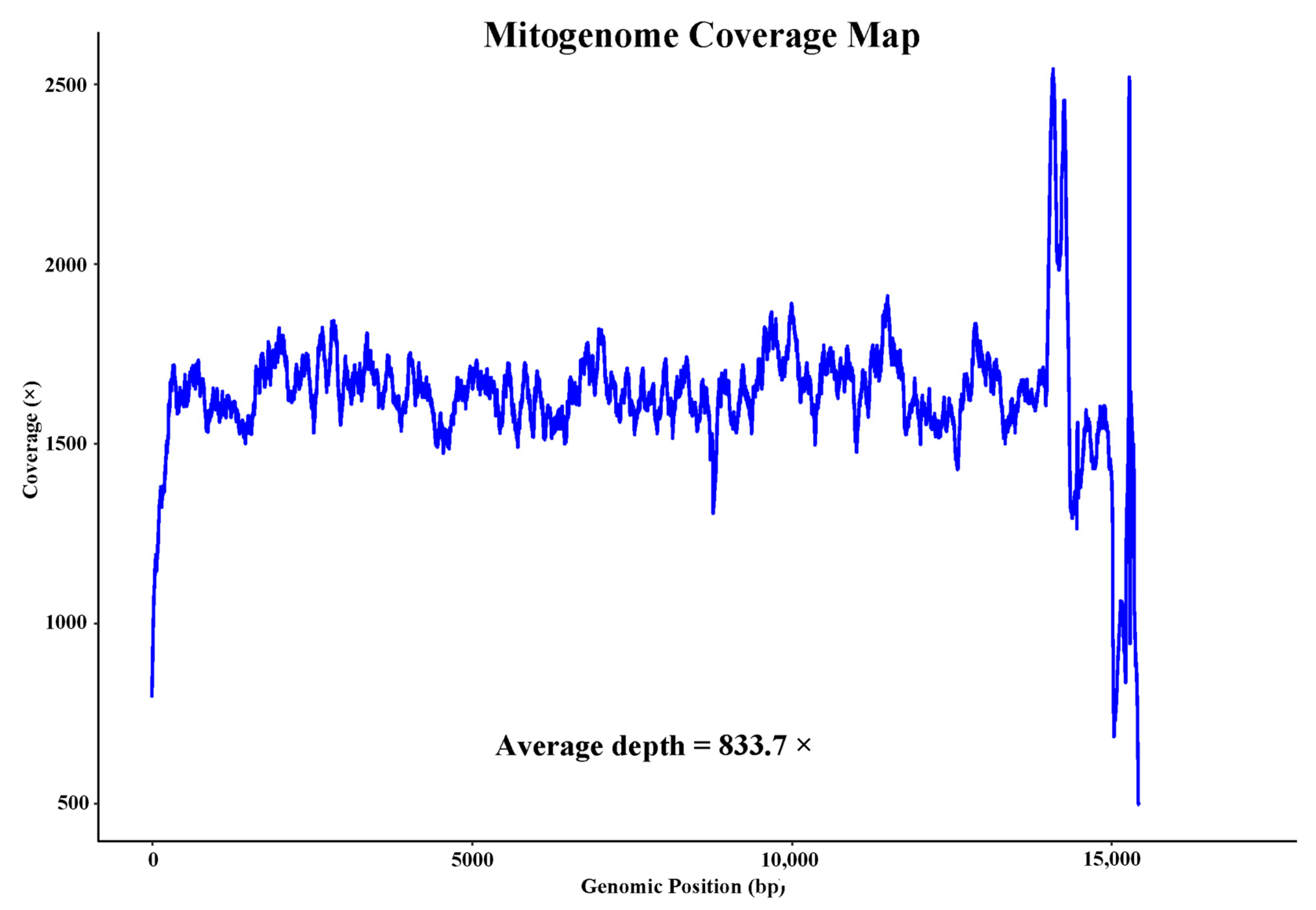
3.1. Genome Characterization and Composition
3.2. Gene Structure and Variation
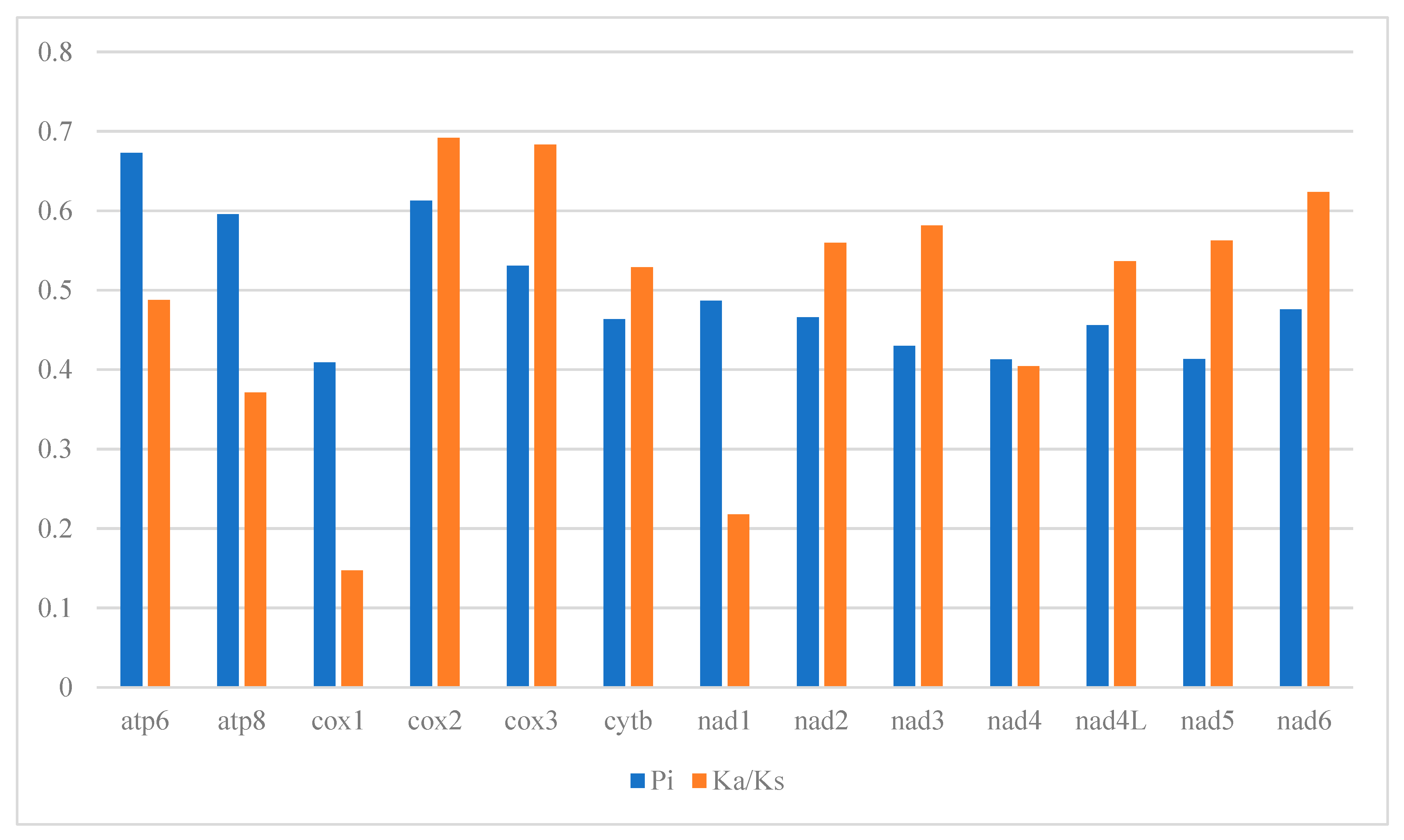
3.3. Protein-Coding Genes Analysis
3.4. Gene Arrangement
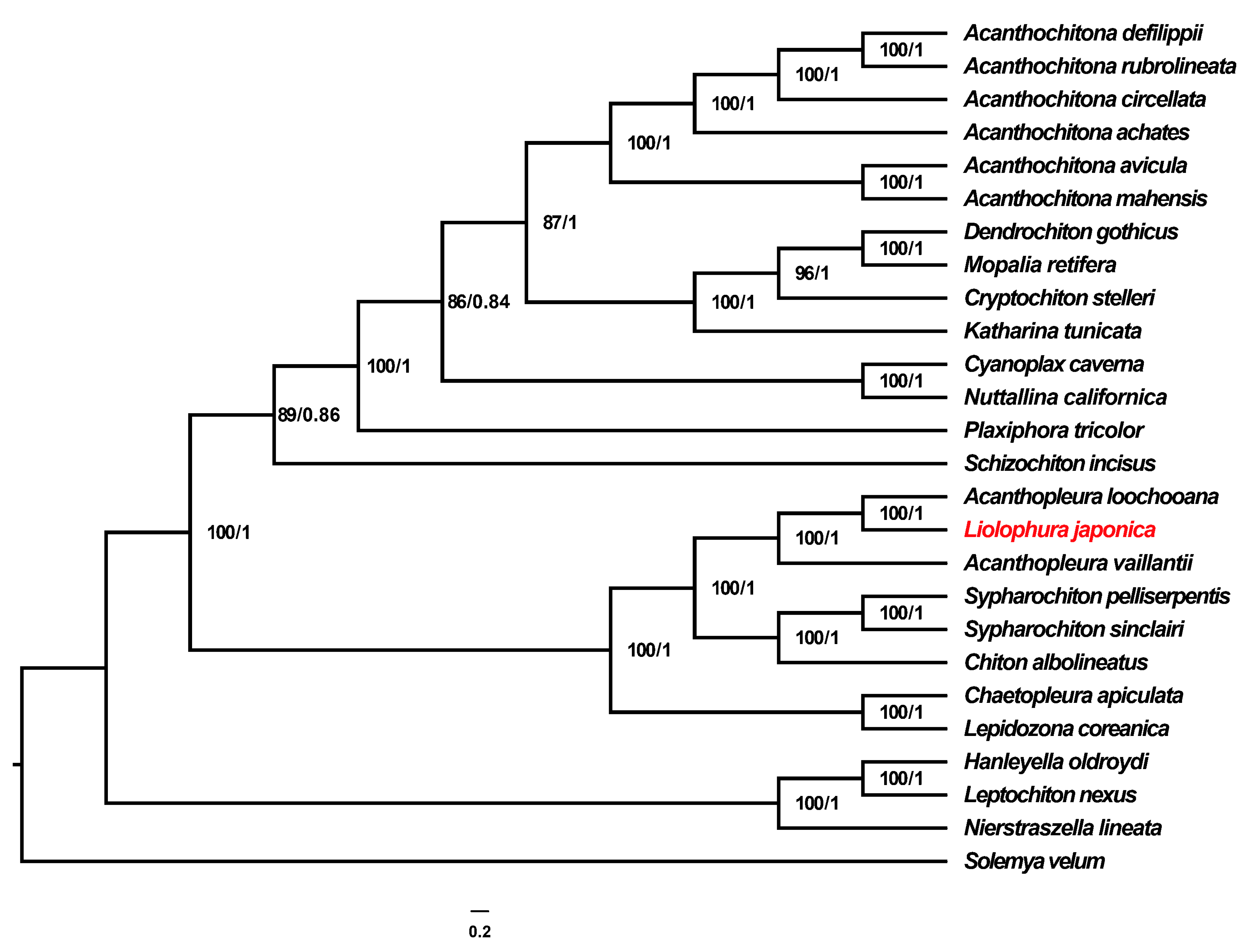
3.5. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Eo, S.H.; DeWoody, J.A. Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proc. R. Soc. Lond. B Biol. Sci. 2010, 277, 3587–3592. [Google Scholar] [CrossRef]
- Anmarkrud, J.A.; Lifjeld, J.T. Complete mitochondrial genomes of eleven extinct or possibly extinct bird species. Mol. Ecol. Resour. 2017, 17, 334–341. [Google Scholar] [CrossRef]
- Irisarri, I.; Uribe, J.E.; Eernisse, D.J.; Zardoya, R. A mitogenomic phylogeny of chitons (Mollusca: Polyplacophora). BMC Evol. Biol. 2020, 20, 1–15. [Google Scholar] [CrossRef]
- Cleary, D.F.R.; Polónia, A.R.M.; Huang, Y.M.; Swierts, T. Compositional variation between high and low prokaryotic diversity coral reef biotopes translates to different predicted metagenomic gene content. Antonie Van Leeuwenhoek 2020, 113, 563–587. [Google Scholar] [CrossRef]
- Suzuki, T.; Furukohri, T.; Okamoto, S. Amino acid sequence of myoglobin from the chiton Liolophura japonica and a phylogenetic tree for molluscan globins. J. Protein Chem. 1993, 12, 45–50. [Google Scholar] [CrossRef]
- Choi, E.H.; Yeo, M.Y.; Kim, G.; Park, B.; Shin, C.R.; Baek, S.Y.; Hwang, U.W. Liolophura species discrimination with geographical distribution patterns and their divergence and expansion history on the northwestern Pacific coast. Sci. Rep. 2021, 11, 17602. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A Toolkit for Animal Mitochondrial Genome Assembly, Annotation and Visualization. Nucleic Acids Res 2019, 47, e63. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ling, C.; Luo, A.; Gao, J. MrBayes 3.2. 6 on Tianhe-1A: A high performance and distributed implementation of phylogenetic analysis. In Proceedings of the 2016 IEEE 22nd International Conference on Parallel and Distributed Systems (ICPADS), Wuhan, China, 13–16 December 2016; IEEE: Piscataway, NJ, USA, 2016; pp. 1181–1186. [Google Scholar]
- Nabholz, B.; Ellegren, H.; Wolf, J.B.W. High Levels of Gene Expression Explain the Strong Evolutionary Constraint of Mitochondrial Protein-Coding Genes. Mol. Biol. Evol. 2013, 30, 272–284. [Google Scholar] [CrossRef]
- Gnad, F.; Forner, F.; Zielinska, D.F.; Birney, E.; Gunawardena, J.; Mann, M. Evolutionary Constraints of Phosphorylation in Eukaryotes, Prokaryotes, and Mitochondria*. Mol. Cell. Proteom. 2010, 9, 2642–2653. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Miao, J.; Li, J.; Ye, Y. Exploring the mitogenomic of Lottiidae (Patellogastropoda): Phylogenetics, gene rearrangement and evolutionary divergence time estimations. BMC Genom. 2024, 25, 1055. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Ren, J.; Liu, X. Mitogenome of the small abalone Haliotis diversicolor Reeve and phylogenetic analysis within Gastropoda. Mar. Genom. 2011, 4, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, C.M.; Pardo-Gandarillas, M.C.; Méndez, M.A.; Sellanes, J.; Sigwart, J.D.; Sirenko, B. Phylogenetic position and morphological descriptions of Chiton species from the south-eastern Pacific. Zool. J. Linn. Soc. 2021, 191, 695–719. [Google Scholar] [CrossRef]
- Sirenko, B. New outlook on the system of chitons (mollusca: Polyplacophora). Venus J. Malacol. Soc. Jpn. 2006, 65, 27–49. [Google Scholar] [CrossRef]
- Liu, X.; Sigwart, J.D.; Sun, J. Phylogenomic analyses shed light on the relationships of chiton superfamilies and shell-eye evolution. Mar. Life Sci. Technol. 2023, 5, 525–537. [Google Scholar] [CrossRef]
- Zhong, Z.; Lan, Y.; Chen, C.; Zhou, Y.; Linse, K.; Li, R.; Sun, J. New mitogenomes in deep-water endemic Cocculinida and Neomphalida shed light on lineage-specific gene orders in major gastropod clades. Front. Ecol. Evol. 2022, 10, 973485. [Google Scholar] [CrossRef]
- Rahuman, S.; Jeena, N.S.; Asokan, P.K.; Vidya, R.; Vijayagopal, P. Mitogenomic architecture of the multivalent endemic black clam (Villorita cyprinoides) and its phylogenetic implications. Sci. Rep. 2020, 10, 15438. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Superfamily | Family | Organism | Length | AT% |
|---|---|---|---|---|---|
| NC_047426.1 | Chitonida | Acanthochitonidae | Acanthochitona avicula Carpenter | 15,203 | 68.6 |
| NC_082305.1 | Chitonida | Acanthochitonidae | Acanthochitona mahensis Winckworth | 15,013 | 67.2 |
| NC_087903.1 | Chitonida | Acanthochitonidae | Acanthochitona defilippii Canefri | 14,999 | 71.7 |
| PQ301026.1 | Chitonida | Acanthochitonidae | Acanthochitona achates Gould | 15,006 | 68.8 |
| PQ301027.1 | Chitonida | Acanthochitonidae | Acanthochitona circellata Reeve | 14,998 | 70.3 |
| PQ301028.1 | Chitonida | Acanthochitonidae | Acanthochitona rubrolineata Lischke | 14,986 | 70.3 |
| KY824658.1 | Chitonida | Chaetopleuridae | Chaetopleura apiculata Say | 15,108 | 72.8 |
| NC_024173.1 | Chitonida | Chitonidae | Sypharochiton sinclairi Gray | 15,028 | 72.2 |
| NC_024174.1 | Chitonida | Chitonidae | Sypharochiton pelliserpentis Quoy & Gaimard | 15,048 | 72.5 |
| NC_047425.1 | Chitonida | Chitonidae | Chiton albolineatus Broderip & Sowerby I | 14,936 | 72.9 |
| NC_068064.1 | Chitonida | Chitonidae | A. loochooana Broderip & Sowerby | 15,366 | 70.1 |
| NC_072326.1 | Chitonida | Chitonidae | L. japonica 1873 | 15,209 | 69.3 |
| NC_082877.1 | Chitonida | Chitonidae | Acanthopleura vaillantii Rochebrune | 15,271 | 67.7 |
| NC_046935.1 | Chitonida | Ischnochitonidae | Lepidozona coreanica Reeve | 16,572 | 70.2 |
| NC_001636.1 | Chitonida | Mopaliidae | Katharina tunicata Wood | 15,532 | 69.4 |
| NC_026850.1 | Chitonida | Mopaliidae | Cryptochiton stelleri Middendorff | 15,082 | 70.5 |
| NC_047424.1 | Chitonida | Mopaliidae | Dendrochiton gothicus Carpenter | 15,288 | 70.3 |
| NC_068065.1 | Chitonida | Mopaliidae | Mopalia retifera Thiele | 14,984 | 70.1 |
| NC_085812.1 | Chitonida | Mopaliidae | Plaxiphora tricolor Thiele | 14,909 | 71 |
| OP994082.1 | Chitonida | Schizochitonidae | Schizochiton incisus Sowerby | 15,615 | 68.7 |
| NC_026848.1 | Chitonida | Tonicellidae | Cyanoplax caverna Eernisse | 15,141 | 74.3 |
| NC_026849.1 | Chitonida | Tonicellidae | Nuttallina californica Reeve | 15,604 | 71 |
| NC_047422.1 | Lepidopleurida | Lepidopleuridae | Leptochiton nexus Carpenter | 15,488 | 68.9 |
| NC_047423.1 | Lepidopleurida | Lepidopleuridae | Hanleyella oldroydi Dall | 15,692 | 68 |
| NC_047421.1 | Lepidopleurida | Nierstraszellidae | Nierstraszella lineata Nierstrasz | 15,765 | 68.9 |
| NC_017612.1 | Solemyida | Solemyidae | Solemya velum Say | 15,660 | 68.1 |
| Locus | Start | Stop | Size (bp) | Start Coding | Stop Coding | Strand |
|---|---|---|---|---|---|---|
| tRNAMet | 1 | 66 | 66 | H | ||
| 12S rRNA | 66 | 928 | 863 | H | ||
| tRNAVal | 919 | 985 | 67 | H | ||
| 16S rRNA | 967 | 2338 | 1372 | H | ||
| tRNALeu | 2281 | 2349 | 69 | H | ||
| tRNALeu | 2362 | 2427 | 66 | H | ||
| nad1 | 2427 | 3378 | 952 | GTG | TAG | H |
| tRNAPro | 3370 | 3435 | 66 | L | ||
| nad6 | 3438 | 3936 | 499 | ATG | TAA | H |
| cob | 3937 | 5077 | 1141 | ATG | TAA | H |
| tRNASer | 5082 | 5150 | 69 | H | ||
| tRNAThr | 5149 | 5216 | 68 | L | ||
| nad4l | 5230 | 5533 | 304 | ATG | TAG | H |
| nad4 | 5526 | 6879 | 1354 | GTG | T | H |
| tRNAHis | 6874 | 6937 | 64 | H | ||
| nad5 | 6937 | 8653 | 1717 | ATG | TAA | H |
| tRNAPhe | 8655 | 8723 | 69 | H | ||
| atp6 | 8759 | 9458 | 700 | ATG | TAG | L |
| atp8 | 9477 | 9639 | 163 | ATG | TAA | L |
| tRNAAsp | 9639 | 9705 | 67 | L | ||
| cox2 | 9703 | 10,396 | 694 | ATG | TAA | L |
| cox1 | 10,404 | 11,937 | 1534 | ATG | TAA | L |
| nad2 | 11,938 | 12,958 | 1021 | ATG | TAG | L |
| tRNASer | 12,961 | 13,029 | 69 | L | ||
| nad3 | 13,027 | 13,384 | 358 | ATG | TAA | L |
| tRNAIle | 13,385 | 13,452 | 68 | L | ||
| tRNAAsn | 13,452 | 13,517 | 66 | L | ||
| tRNAArg | 13,528 | 13,588 | 61 | L | ||
| tRNAAla | 13,599 | 13,665 | 67 | L | ||
| tRNALys | 13,661 | 13,727 | 67 | L | ||
| cox3 | 13,754 | 14,534 | 781 | ATG | TAA | L |
| tRNAGlu | 14,728 | 14,794 | 67 | H | ||
| tRNAGly | 14,807 | 14,876 | 70 | H | ||
| tRNAGln | 14,924 | 14,993 | 70 | H | ||
| tRNATrp | 14,993 | 15,059 | 67 | H | ||
| tRNATyr | 15,065 | 15,129 | 65 | H | ||
| tRNAPro | 15,129 | 15,191 | 63 | H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Liu, Z.; Dong, W.; Xing, B.; Luo, S.; Xiang, P. Deciphering the First Mitochondrial Genome of the Liolophura Pilsbry, 1893 Genus: An Extensive Phylogenetic Study Within the Chitonidae Family. Genes 2025, 16, 606. https://doi.org/10.3390/genes16050606
Zhou Q, Liu Z, Dong W, Xing B, Luo S, Xiang P. Deciphering the First Mitochondrial Genome of the Liolophura Pilsbry, 1893 Genus: An Extensive Phylogenetic Study Within the Chitonidae Family. Genes. 2025; 16(5):606. https://doi.org/10.3390/genes16050606
Chicago/Turabian StyleZhou, Qianqian, Zhiyong Liu, Weifeng Dong, Bingpeng Xing, Site Luo, and Peng Xiang. 2025. "Deciphering the First Mitochondrial Genome of the Liolophura Pilsbry, 1893 Genus: An Extensive Phylogenetic Study Within the Chitonidae Family" Genes 16, no. 5: 606. https://doi.org/10.3390/genes16050606
APA StyleZhou, Q., Liu, Z., Dong, W., Xing, B., Luo, S., & Xiang, P. (2025). Deciphering the First Mitochondrial Genome of the Liolophura Pilsbry, 1893 Genus: An Extensive Phylogenetic Study Within the Chitonidae Family. Genes, 16(5), 606. https://doi.org/10.3390/genes16050606