

Signaling Pathways in Gliomas

Abstract
:1. Introduction
2. Signaling Pathways in Oncogenesis
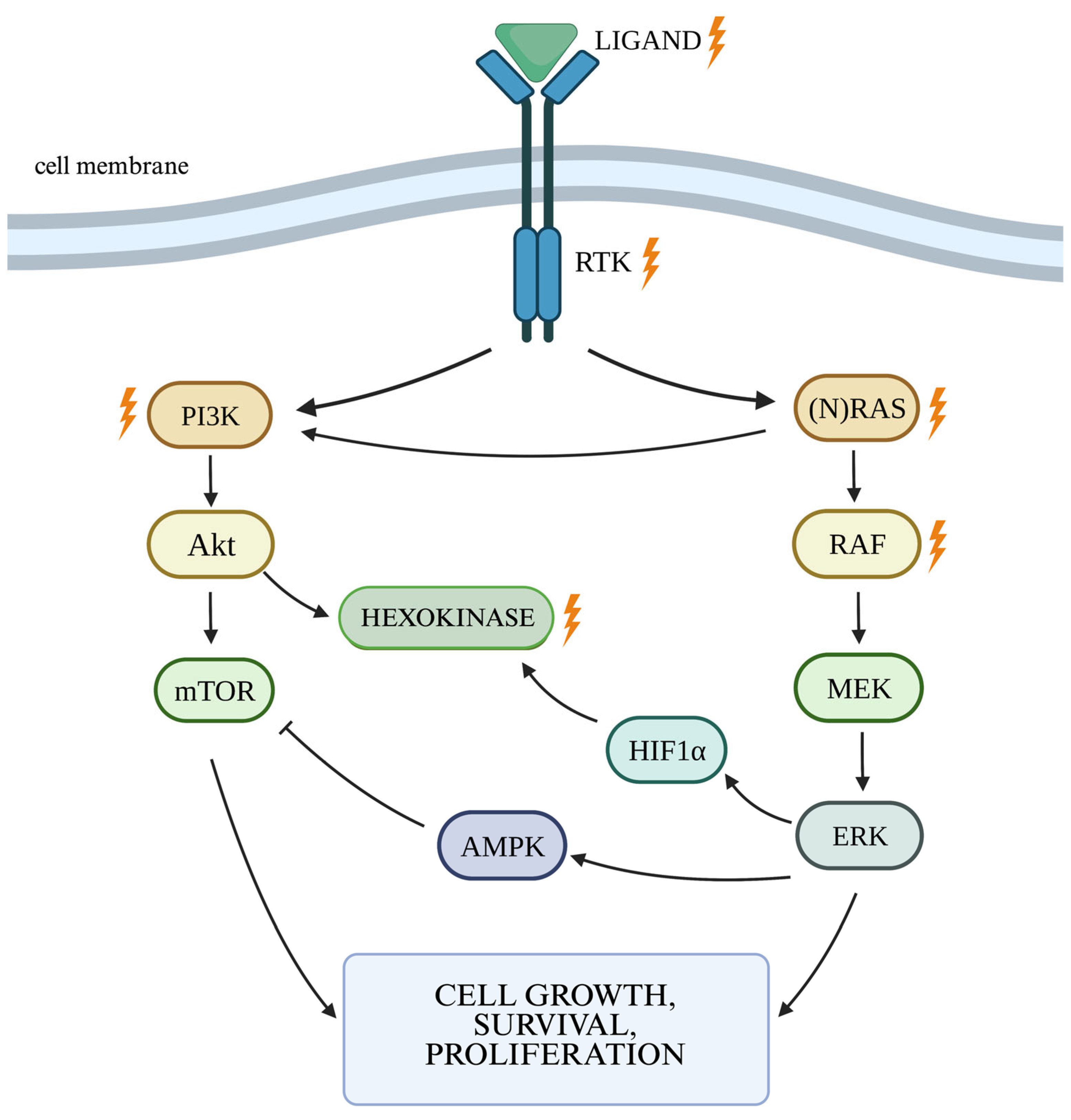
2.1. RAS/MAPK/ERK Cellular Pathway
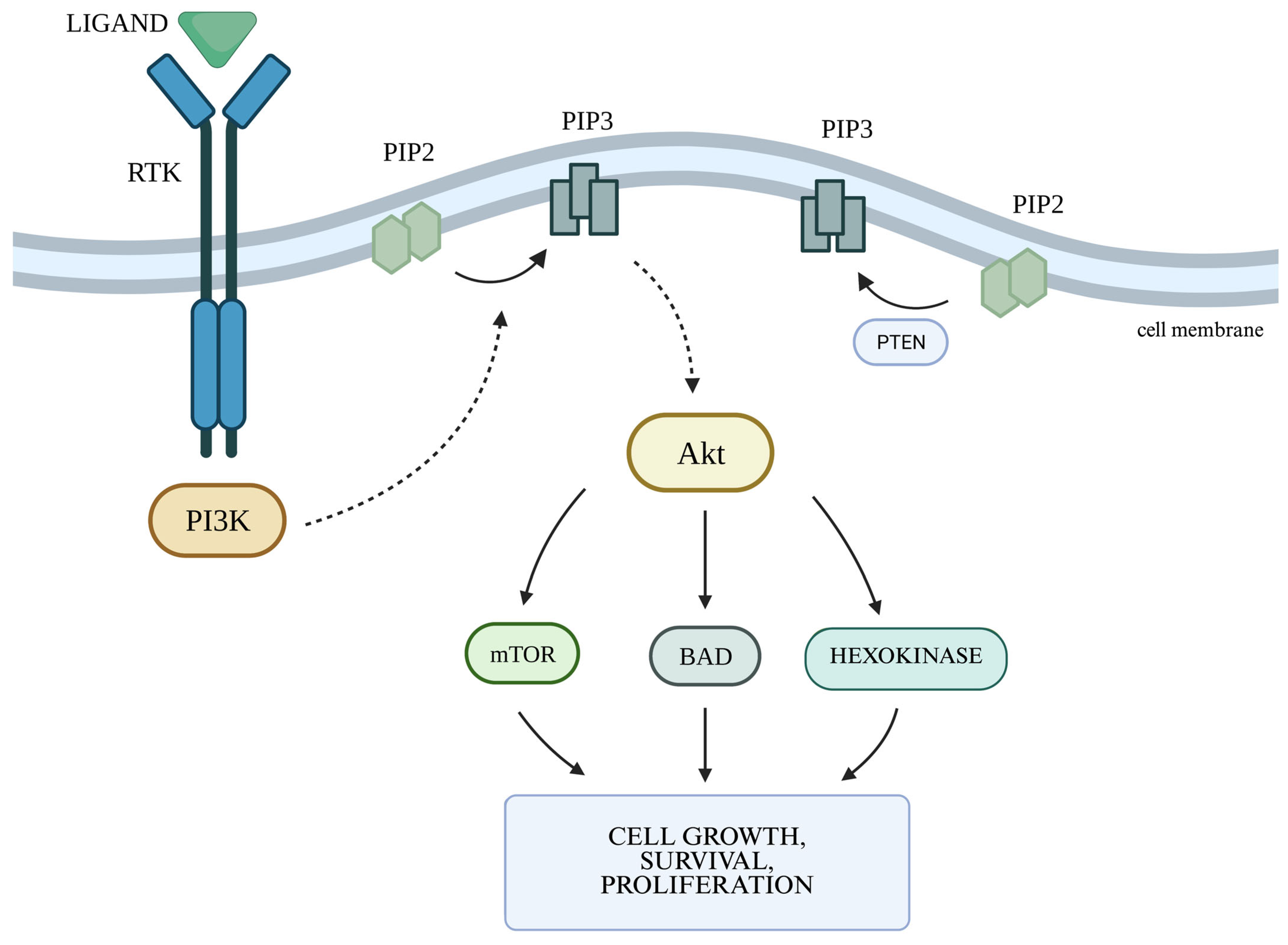
2.2. The PI3K/Akt Cellular Pathway and the Autophagy Process
2.3. Other Signaling Pathways in Cancerogenesis
3. Enzymes in Oncogenesis
3.1. Isocitrate Dehydrogenase
3.2. MGMT Promoter Methylation
3.3. Poly-ADP-Ribose Polymerases
3.4. Hexokinase 2
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GBM | glioblastoma multiforme |
| WHO | World Health Organization |
| CNS | central nervous system |
| IDH | isocitrate dehydrogenase |
| RTK | receptor protein with a tyrosine kinase function |
| RAS | rat sarcoma |
| RAF | rapidly accelerated fibrosarcoma |
| MEK | mitogen-activated kinase |
| ERK | extracellular signal-regulated kinase |
| EGFR | epidermal growth factor receptor |
| EGF | epidermal growth factor |
| TGF-α | transforming growth factor α |
| HIF1 | hypoxia-inducible factor 1 |
| GDP | guanosine diphosphate |
| GTP | guanosine triphosphate |
| PIP3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PIP2 | phosphatidylinositol (4,5)-bisphosphate |
| PKB | protein kinase B |
| BAD | Bcl-2-associated death promoter |
| mTOR | serine/threonine kinase mammalian target of rapamycin |
| mTORC | mTOR complex |
| ATG | autophagy-related genes |
| PI3KC3 | the third class PI3K complex |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α |
| PIK3R1 | phosphatidylinositol-4,5-bisphosphate 3-kinase regulatory subunit 1 |
| HK2 | hexokinase 2 |
| IL-6 | interleukin 6 |
| JAK/STAT3 | Janus kinase/signal transducer activator of transcription protein |
| PTCH | patched |
| SMO | smoothened |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NADH | reduced form of nicotinamide adenine dinucleotide |
| D-2HG | D-2-hydroxyglutarate |
| TET | ten–eleven translocation |
| GLUT1 | glucose transporter 1 |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| NMDA | N-methyl-D-aspartate |
| OS | overall survival |
| PFS | progression-free survival |
| O6-BG | O6-benzylguanine |
| miR-198 | microRNA 198 |
| PARP | poly-ADP-ribose polymerases |
| SL | synthetic lethality |
| ATP | adenosine triphosphate |
| IGF | insulin-like growth factor-1 |
References
- Louis, D.N.; Perry, A. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Leece, R.; Xu, J. Global incidence of malignant brain and other central nervous system tumors by histology, 2003–2007. Neuro Oncol. 2017, 19, 1553–1564. [Google Scholar] [CrossRef]
- Marenco-Hillembrand, L.; Wijesekera, O. Trends in glioblastoma: Outcomes over time and type of intervention: A systematic evidence based analysis. J. Neurooncol 2020, 147, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Fisher, J.L.; Schwartzbaum, J.A. Epidemiology of brain tumors. Neurol. Clin. 2007, 25, 867–890. [Google Scholar] [CrossRef] [PubMed]
- Scheurer, M.E.; Etzel, C.J. Familial aggregation of glioma: A pooled analysis. Am. J. Epidemiol. 2010, 172, 1099–1107. [Google Scholar] [CrossRef]
- Claus, E.B.; Walsh, K.M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus. 2015, 38, E6. [Google Scholar] [CrossRef]
- Amirian, E.S.; Zhou, R. Approaching a scientific consensus on the association between allergies and glioma risk: A report from the Glioma International Case–Control Study. Cancer Epidemiol. Biomark. Prev. 2016, 25, 282–290. [Google Scholar] [CrossRef]
- Vienne-Jumeau, A.; Tafani, C. Environmental risk factors of primary brain tumors: A review. Rev. Neurol. 2019, 175, 664–678. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.H.; Farma, J.M. Cutaneous Melanoma: Etiology and Therapy, 1st ed.; Codon Publications: Brisbane, Australia, 2017; pp. 3–23. [Google Scholar]
- Dullea, A.; Marignol, L. MGMT testing allows for personalised therapy in the temozolomide era. Tumour Biol. 2016, 37, 87–96. [Google Scholar] [CrossRef]
- An Investigational Immunotherapy Study of Temozolomide Plus Radiation Therapy with Nivolumab or Placebo, for Newly Diagnosed Patients with Glioblastoma (GBM, a Malignant Brain Cancer) (CheckMate548). Available online: https://clinicaltrials.gov/ct2/show/NCT02667587 (accessed on 9 February 2023).
- Filley, A.C.; Henriquez, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kim, T.M. Treatment with pembrolizumab in programmed death ligand 1–positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE–028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef]
- Stachyra-Strawa, P.; Ciesielka, M. The role of immunotherapy and molecular-targeted therapy in the treatment of melanoma. Oncol. Rep. 2021, 46, 158. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Karsy, M.; Guan, J. New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr. Neurol. Neurosci. Rep. 2017, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Robinson, J.P.; VanBrocklin, M.W. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene 2010, 29, 335–344. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK signaling pathways in cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef] [PubMed]
- Oprita, A.; Baloi, S.C. Updated Insights on EGFR Signaling Pathways in Glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Roth, P.; Weller, M. Challenges to targeting epidermal growth factor receptor in glioblastoma: Escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014, 8, 4–9. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Alexandru, O.; Purcaru, S.O. The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma. Int. J. Mol. Sci. 2018, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Rhun, E.L.; Preusser, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Bolcaen, J.; Nair, S. Novel Receptor Tyrosine Kinase Pathway Inhibitors for Targeted Radionuclide Therapy of Glioblastoma. Pharmaceuticals 2021, 14, 626. [Google Scholar] [CrossRef]
- Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct. Funct. 2001, 26, 25–35. [Google Scholar] [CrossRef]
- Samer, N.; Abdellatef, S. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. [Google Scholar]
- Zhou, K.L.; Zhu, Z.H. Increased hexokinase-2 as a novel biomarker for the diagnosis and correlating with disease severity in rheumatoid arthritis. Medecine 2021, 100, e26504. [Google Scholar] [CrossRef] [PubMed]
- Seyedmirzaei, H.; Shobeiri, P. VEGF levels in patients with glioma: A systematic review and meta-analysis. Rev. Neurosci. 2020, 32, 191–202. [Google Scholar] [CrossRef]
- Osterberg, N.; Ferrara, N. Decrease of VEGF-A in myeloid cells attenuates glioma progression and prolongs survival in an experimental glioma model. Neuro Oncol. 2016, 18, 939–949. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Yuan, L. KRAS gene polymorphisms are associated with the risk of glioma: A two-center case-control study. Transl. Pediatr. 2021, 10, 579–586. [Google Scholar] [CrossRef]
- Figueras, A.; Arbos, M.A. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer 2013, 13, 125. [Google Scholar] [CrossRef]
- Holmen, S.L.; Williams, B.O. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005, 65, 8250–8255. [Google Scholar] [CrossRef]
- Schittenhelm, J.; Krischker, N. Oncogenic KRAS hotspot mutations are rare in IDH-mutant gliomas. Brain Pathol. 2019, 29, 321–324. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Fuller, G.N. Retrospective analysis of molecular and Immunohistochemical characterization of 381 primary brain tumors. J. Neuropathol. Exp. Neurol. 2017, 76, 179–188. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Floris, P.B. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Arakawa, Y. Infrequent RAS mutation is not associated with specific histological phenotype in gliomas. BMC Cancer 2021, 21, 1025. [Google Scholar] [CrossRef]
- Mc Grail Fernandez, K.A. Targeting NRAS Mutant Melanomas Through Metabolic Stress. Ph.D. Thesis, Universidad Autonoma de Barcelona, Barcelona, Spain, 2021. [Google Scholar]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Burke, J.E. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.; Dyczynski, M. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Tanaka, S.; Batchelor, T.T.; Iafrate, A.J.; Dias-Santagata, D.; Borger, D.R.; Ellisen, L.W.; Yang, D.; Louis, D.N.; Cahill, D.P.; Chi, A.S. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol. Commun. 2019, 7, 66. [Google Scholar] [CrossRef]
- Ligresti, G.; Militello, L. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.H.; Lauring, J. Recurrent AKT mutations in human cancers: Functional consequences and effects on drug sensitivity. Oncotarget 2016, 7, 4241–4251. [Google Scholar] [CrossRef]
- Cheng, C.K.; Fan, Q.W. PI3K signaling in glioma-animal models and therapeutic challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hu, R. PTEN gene mutations correlate to poor prognosis in glioma patients: A meta-analysis. Onco Targets Ther. 2016, 9, 3485–3492. [Google Scholar] [PubMed]
- Lefranc, F.; Facchini, V. Proautophagic drugs: A novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 2007, 12, 1395–1403. [Google Scholar] [CrossRef]
- Zhuo, B.; Li, Y. PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem. Biophys. Res. Commun. 2015, 464, 401–406. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L. Activation of a metabolic gene regulatory network downstream of mTOR complex. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Vartanian, A.; Agnihotri, S. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget 2016, 7, 69518–69535. [Google Scholar] [CrossRef]
- Qiu, M.Z.; Han, B. Expression of hypoxia-inducible factor-1α and hexokinase-II in gastric adenocarcinoma: The impact on prognosis and correlation to clinicopathologic features. Tumour Biol. 2011, 32, 159–166. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53- deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Shulga, N. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef]
- Kao, G.D.; Jiang, Z. Inhibition of phosphatidylinositol–3–OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar]
- Li, Y.J.; Lei, Y.H. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Pasquier, B. SAR405, a PIK3C3/VPS34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Marin-Muller, C. Mesothelin confers pancreatic cancer cell resistance to TNF-alpha-induced apoptosis through Akt/PI3K/NF-kappaB activation and IL-6/Mcl-1 overexpression. Mol. Cancer 2011, 10, 106. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S. The varied roles of notch in cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Parmigiani, E. Interferon-γ resistance and immune evasion in glioma develop via Notch-regulated co-evolution of malignant and immune cells. Dev. Cell 2022, 57, 1847–1865. [Google Scholar] [CrossRef]
- Hassan, K.A. Small cell lung cancer heterogeneity: Elevated a Notch above the Rest. J. Thorac. Dis. 2018, 10, 554–556. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic. Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Song, P.; Gao, Z. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol. 2024, 17, 46. [Google Scholar] [CrossRef]
- Picca, A.; Berzero, G. The clinical use of IDH1 and IDH2 mutations in gliomas. Expert Rev. Mol. Diagn. 2018, 18, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Krell, D.; Assoku, M. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH Mutations in Glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastomamultiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Zhang, L.; Sorensen, M.D. D-2-hydroxyglutarate is an intercellular mediator in IDH-Mutant gliomas inhibiting complement and T cells. Clin. Cancer Res. 2018, 24, 5381–5391. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- McFaline-Figueroa, J.R. Brain Tumors. Am. J. Med. 2018, 131, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Zawlik, I.; Vaccarella, S. Promoter methylation and polymorphisms of the MGMT gene in glioblastomas: A population-based study. Neuroepidemiology 2009, 32, 21–29. [Google Scholar] [CrossRef]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neurooncol 2017, 134, 505–512. [Google Scholar] [CrossRef]
- Christians, A.; Adel-Horowski, A. The prognostic role of IDH mutations in homogeneously treated patients with anaplastic astrocytomas and glioblastomas. Acta Neuropathol. Commun. 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Gusyatiner, O.; Hegi, M.E. Glioma epigenetics: From subclassification to novel treatment options. Semin. Cancer Biol. 2018, 51, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.H.; Zhang, P. Association of MGMT Promoter Methylation Status With Survival Outcomes in Patients With High-Risk Glioma Treated With Radiotherapy and Temozolomide: An Analysis From the NRG Oncology/RTOG 0424 Trial. JAMA Oncol. 2018, 4, 1405–1409. [Google Scholar] [CrossRef]
- Fleming, J.; McElroy, J. Elevated MGMT Gene Expression is Independently Associated with Worse Overall Survival in NRG Oncology/RTOG 9813: A Phase III Study of Radiation Therapy (RT) and Temozolomide (TMZ) vs. RT and Nitrosourea (NU) in Anaplastic Grade III Glioma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, S46–S47. [Google Scholar] [CrossRef]
- Jungk, C.; Chatziaslanidou, D. Chemotherapy with BCNU in recurrent glioma: Analysis of clinical outcome and side effects in chemotherapy-naïve patients. BMC Cancer 2016, 16, 81. [Google Scholar] [CrossRef]
- Quinn, J.A.; Jiang, S.X. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef]
- Nie, E.; Jin, X. MiR-198 enhances temozolomide sensitivity in glioblastoma by targeting MGMT. J. Neurooncol. 2017, 133, 59–68. [Google Scholar] [CrossRef]
- Dębska, S.; Kubicka, J. Inhibitory PARP—Podstawy teoretyczne i zastosowanie kliniczne. Postep. Hig. Med. Dosw. 2012, 66, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Xavier, M.-A.; Rezende, F. BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme. Biomolecules 2021, 11, 1188. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Stachyra-Strawa, P.; Cisek, P. The role of hexokinase in cancer. Postep. Hig. Med. Dosw. 2020, 74, 144–150. [Google Scholar] [CrossRef]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef]
- Deeb, S.S.; Malkki, M. Human hexokinase II: Sequence and homology to other hexokinases. Biochem. Biophys. Res. Commun. 1993, 197, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.; Ko, Y.; Pedersen, P. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Nederlof, R.; Gürel-Gurevin, E. Reducing mitochondrial bound hexokinase II mediates transition from non-injurious into injurious ischemia/reperfusion of the intact heart. J. Physiol. Biochem. 2016, 73, 323–333. [Google Scholar] [CrossRef]
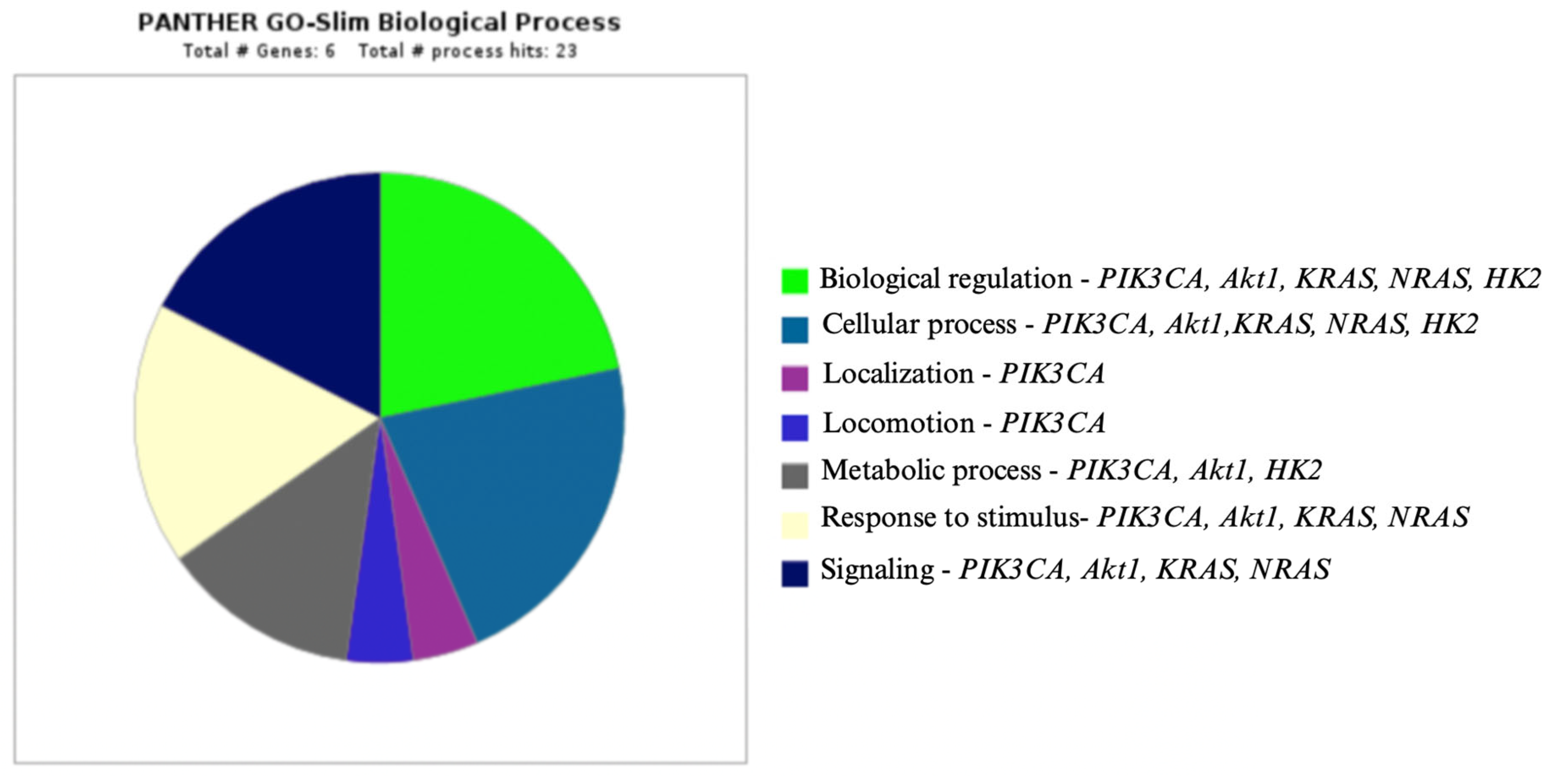
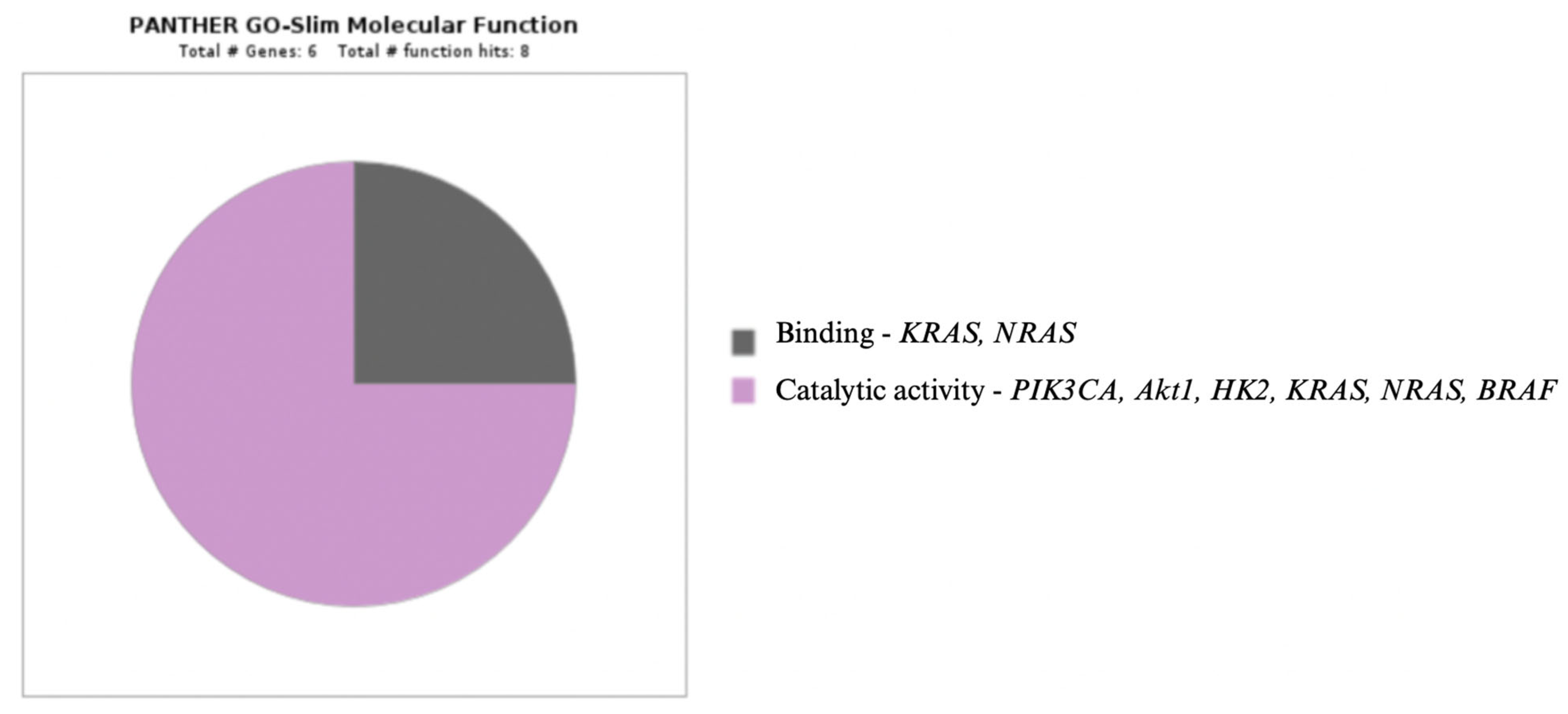
- Panther Classification System. Available online: http://www.pantherdb.org (accessed on 22 December 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GLIOMA TYPE | POSSIBLE GRADES OF MALIGNANCY ACCORDING TO WHO |
|---|---|
| Astrocytoma IDH-mutant | 2, 3, 4 |
| Oligodendroglioma IDH-mutant, 1p/19q codeletion | 2, 3 |
| Glioblastoma multiforme IDH-wildtype | 4 |
| IDH-WILDTYPE | IDH-MUTANT | |
|---|---|---|
| IDH MUTATION | No | Yes |
| FREQUENCY OF OCCURRENCE | More common in gliomas G4 (>90% GBM) | More common in gliomas G2 and G3 |
| ONSET | Primary | Secondary |
| LOCATION | Subtentorially | Frontal lobe |
| AVERAGE AGE OF ONSET | ~62 years of age | ~44 years of age |
| PROGNOSIS | Worse | Better |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachyra, P.; Grzybowska-Szatkowska, L. Signaling Pathways in Gliomas. Genes 2025, 16, 600. https://doi.org/10.3390/genes16050600
Stachyra P, Grzybowska-Szatkowska L. Signaling Pathways in Gliomas. Genes. 2025; 16(5):600. https://doi.org/10.3390/genes16050600
Chicago/Turabian StyleStachyra, Paulina, and Ludmiła Grzybowska-Szatkowska. 2025. "Signaling Pathways in Gliomas" Genes 16, no. 5: 600. https://doi.org/10.3390/genes16050600
APA StyleStachyra, P., & Grzybowska-Szatkowska, L. (2025). Signaling Pathways in Gliomas. Genes, 16(5), 600. https://doi.org/10.3390/genes16050600