
Combined Analysis of Transcriptomes and Metabolomes Reveals That MeJA-Mediated Flavonoid Biosynthesis Is Crucial for Pigment Deposition in Naturally Colored Green Cotton Fibers
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
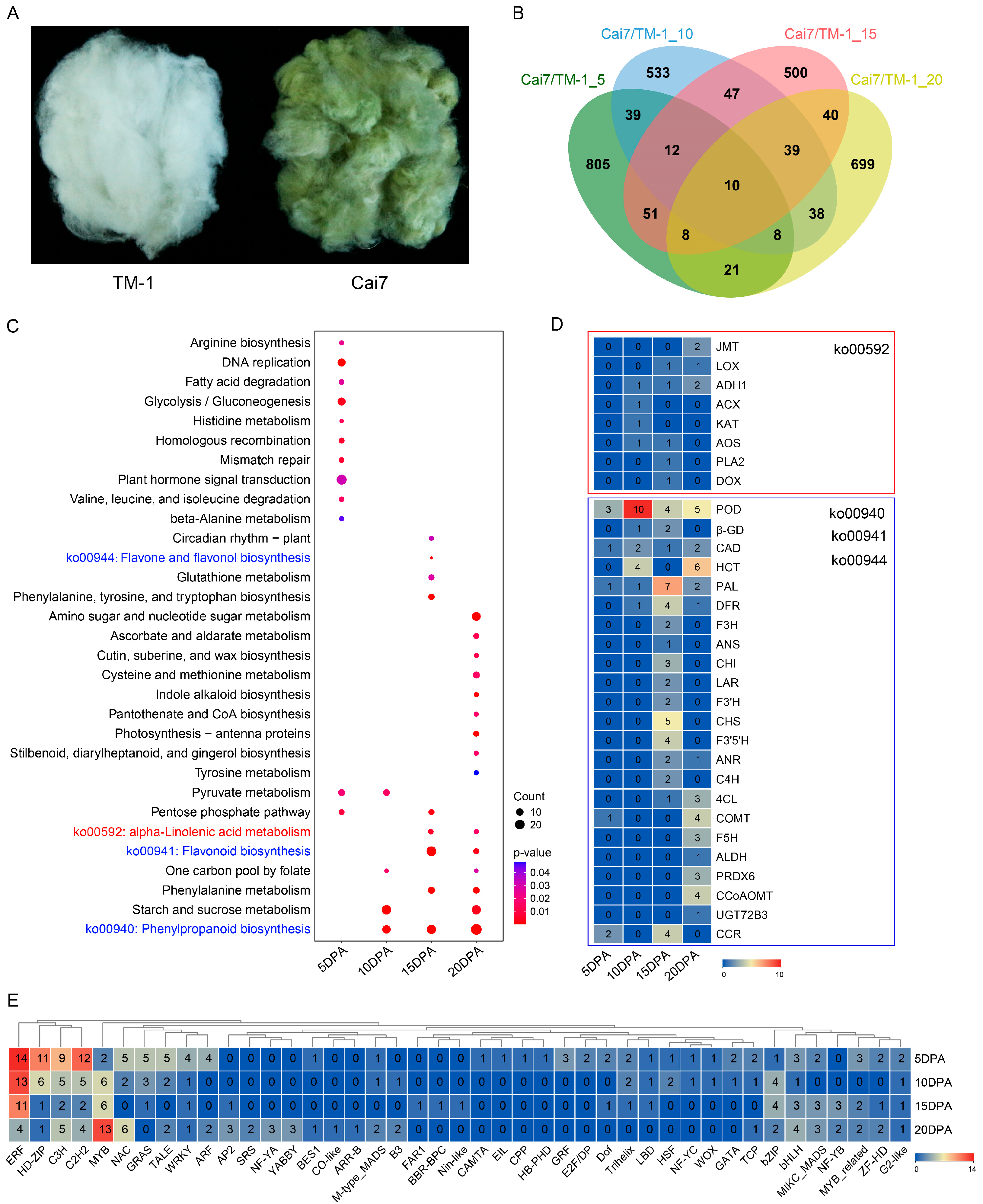
2.1. Comparative Analysis of the Transcriptomes Between Green and White Fibers
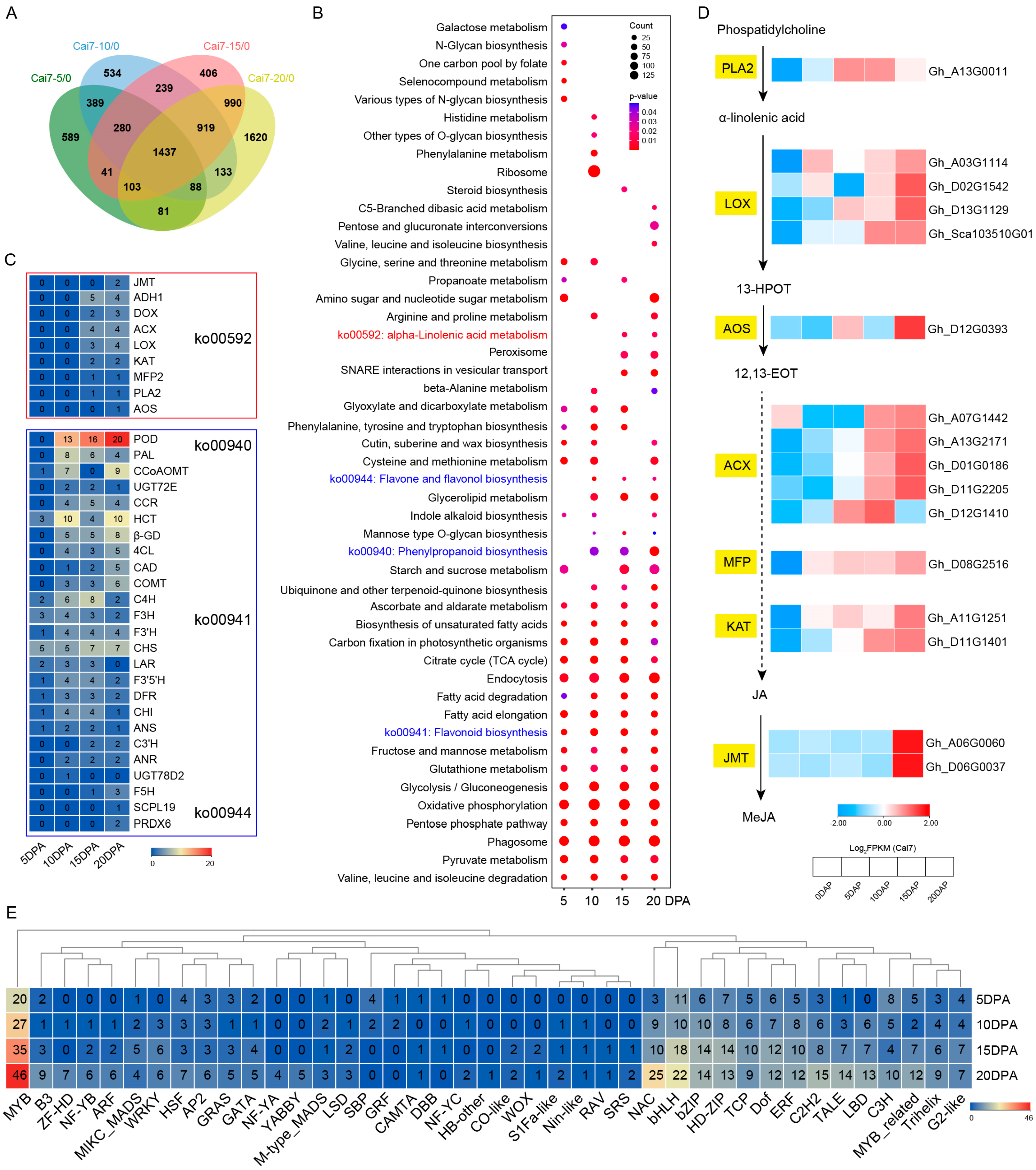
2.2. Transcriptomic Profiling of GCFs Across Developmental Stages
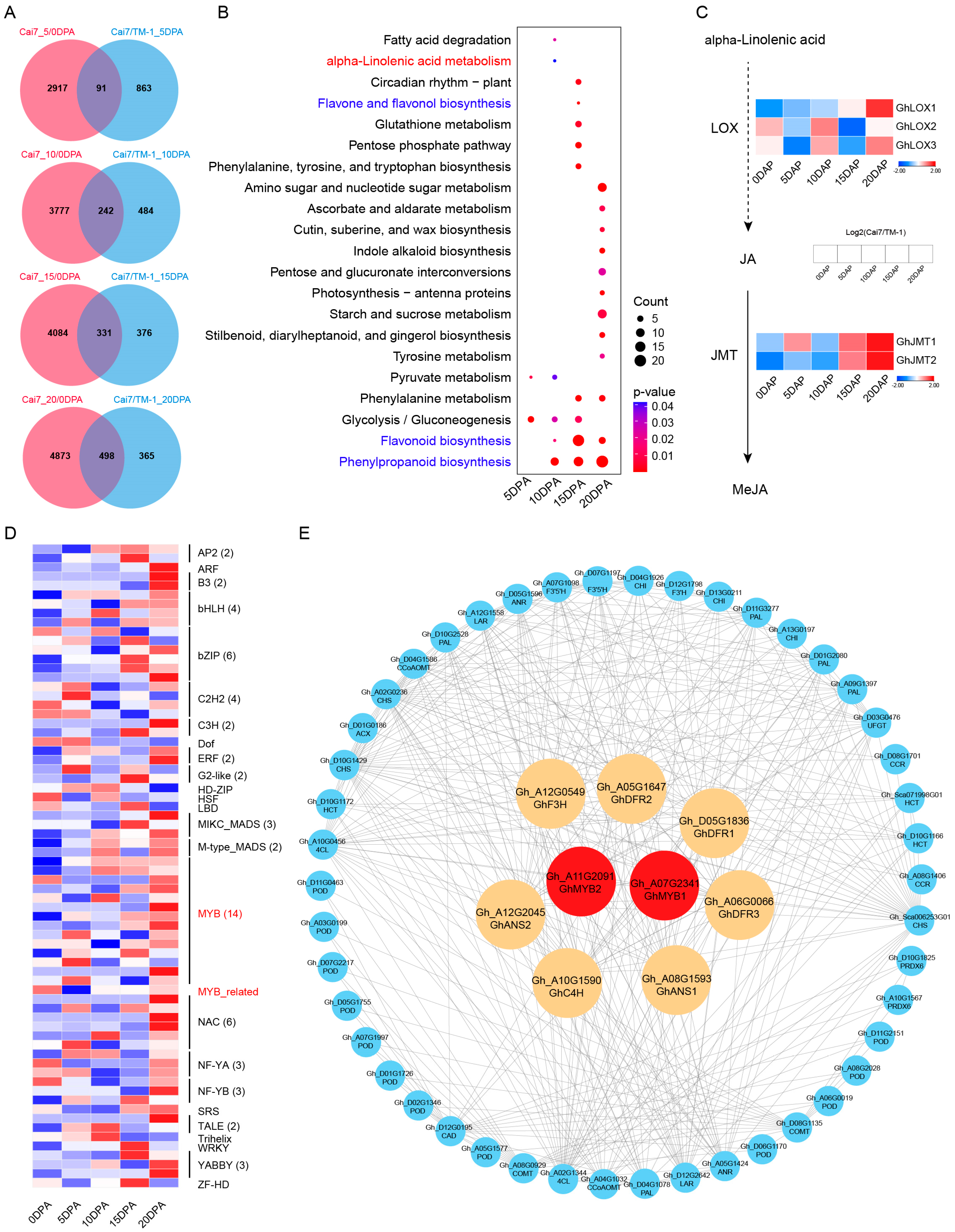
2.3. Multi-Dimensional Conjoint Analysis of White and Green Fibers Transcriptomes
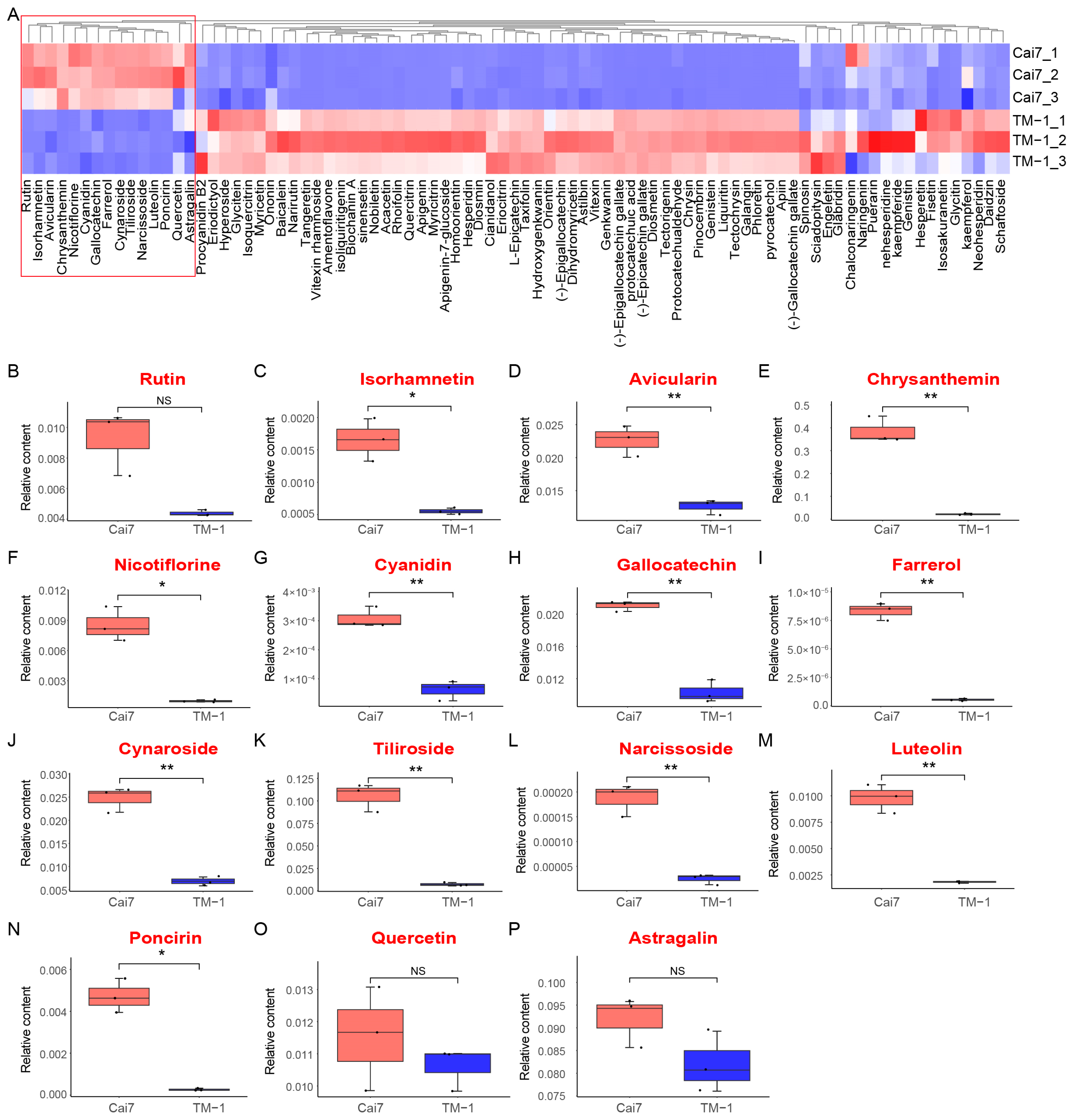
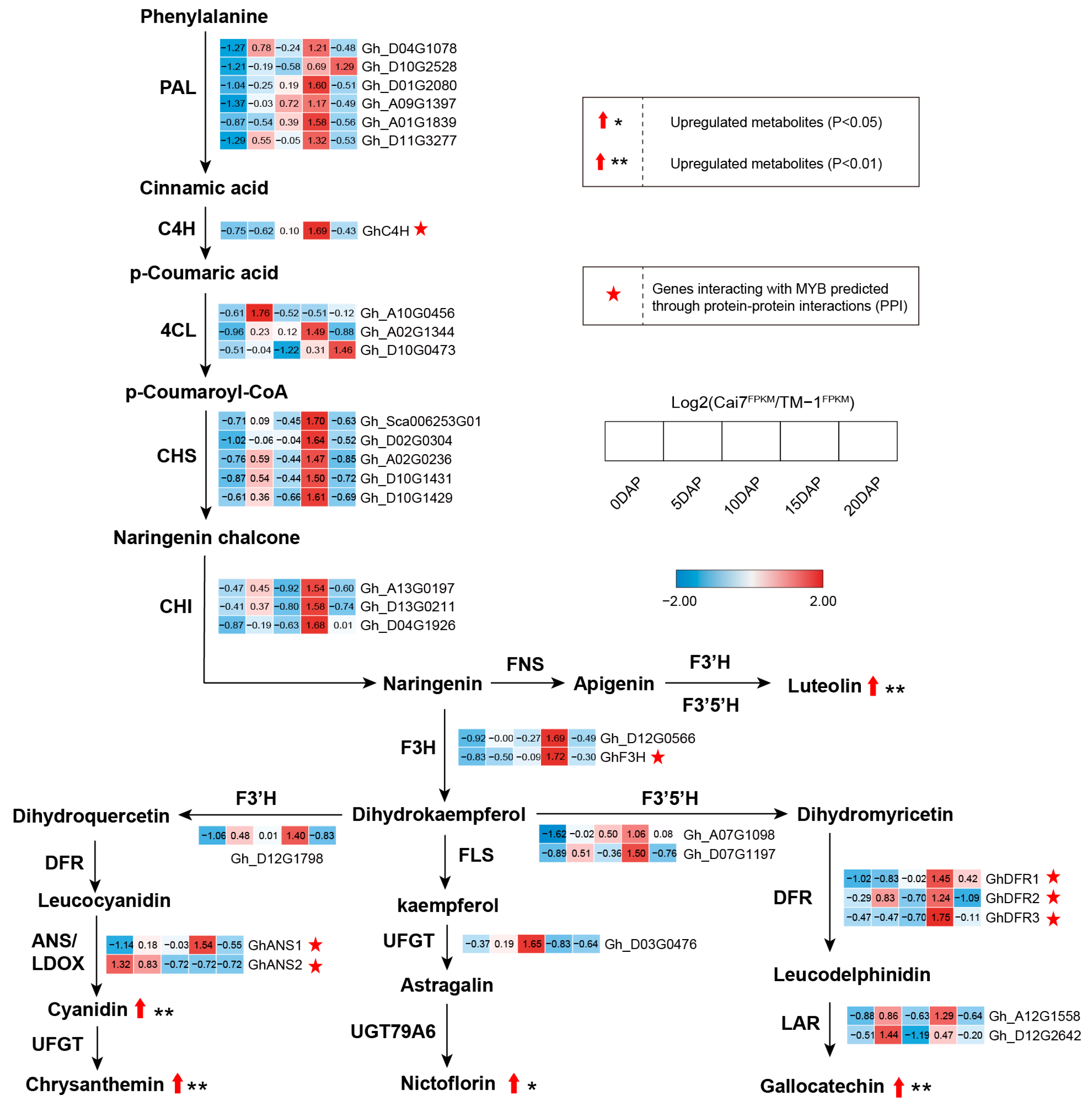
2.4. Comparative Metabolomic Profiling of White Versus Green Fibers
2.5. Combined Analysis of Transcriptome and Metabolome in White and Green Fiber Variants
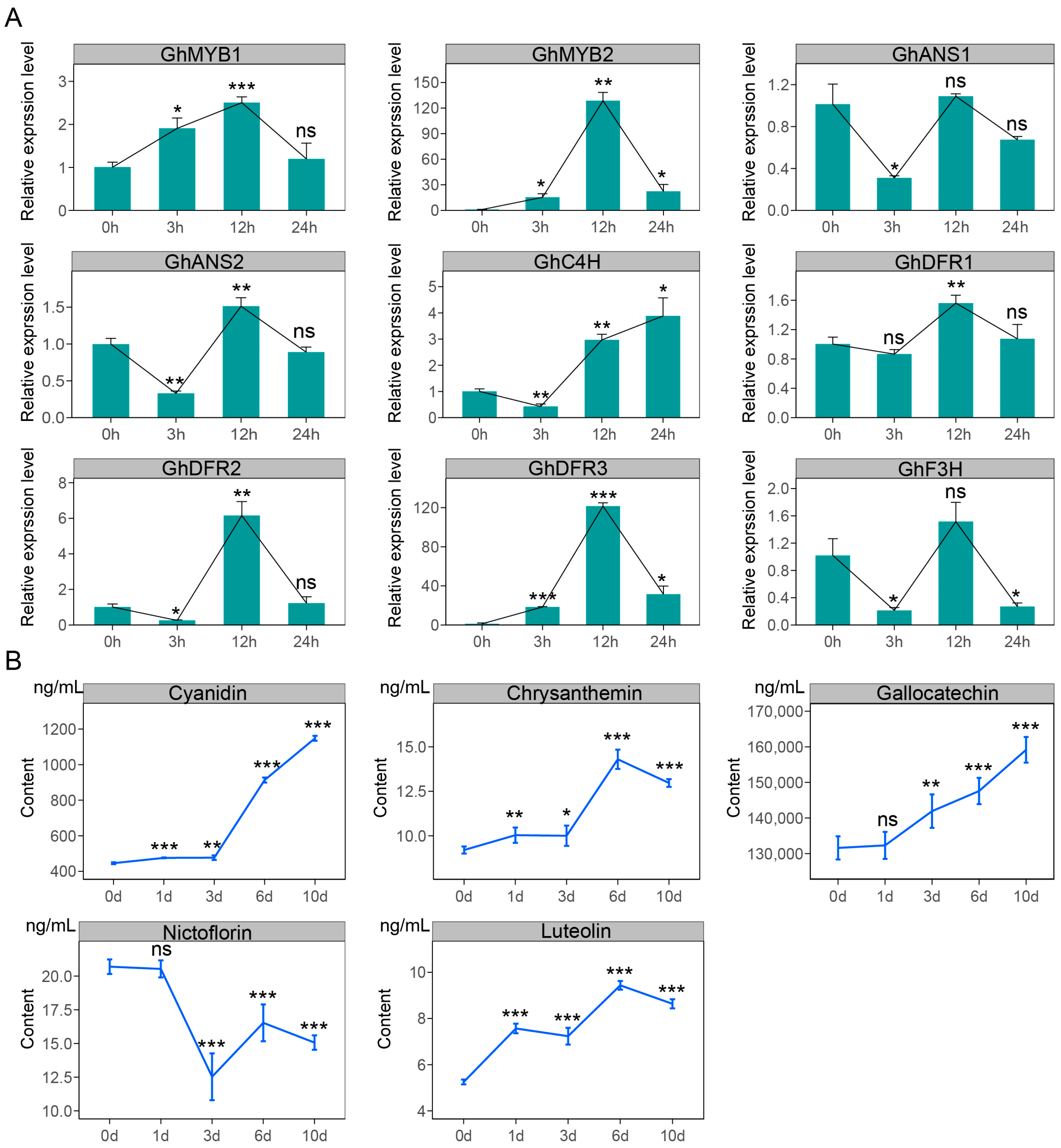
2.6. Changes in Flavonoid Pathway Gene Expression Levels and Metabolite Contents in GCFs Under Exogenous MeJA Treatment
3. Discussion
3.1. α-Linolenic Acid Metabolism and Flavonoid-Related Pathways Are Significantly Enriched During Green Cotton Fiber Development
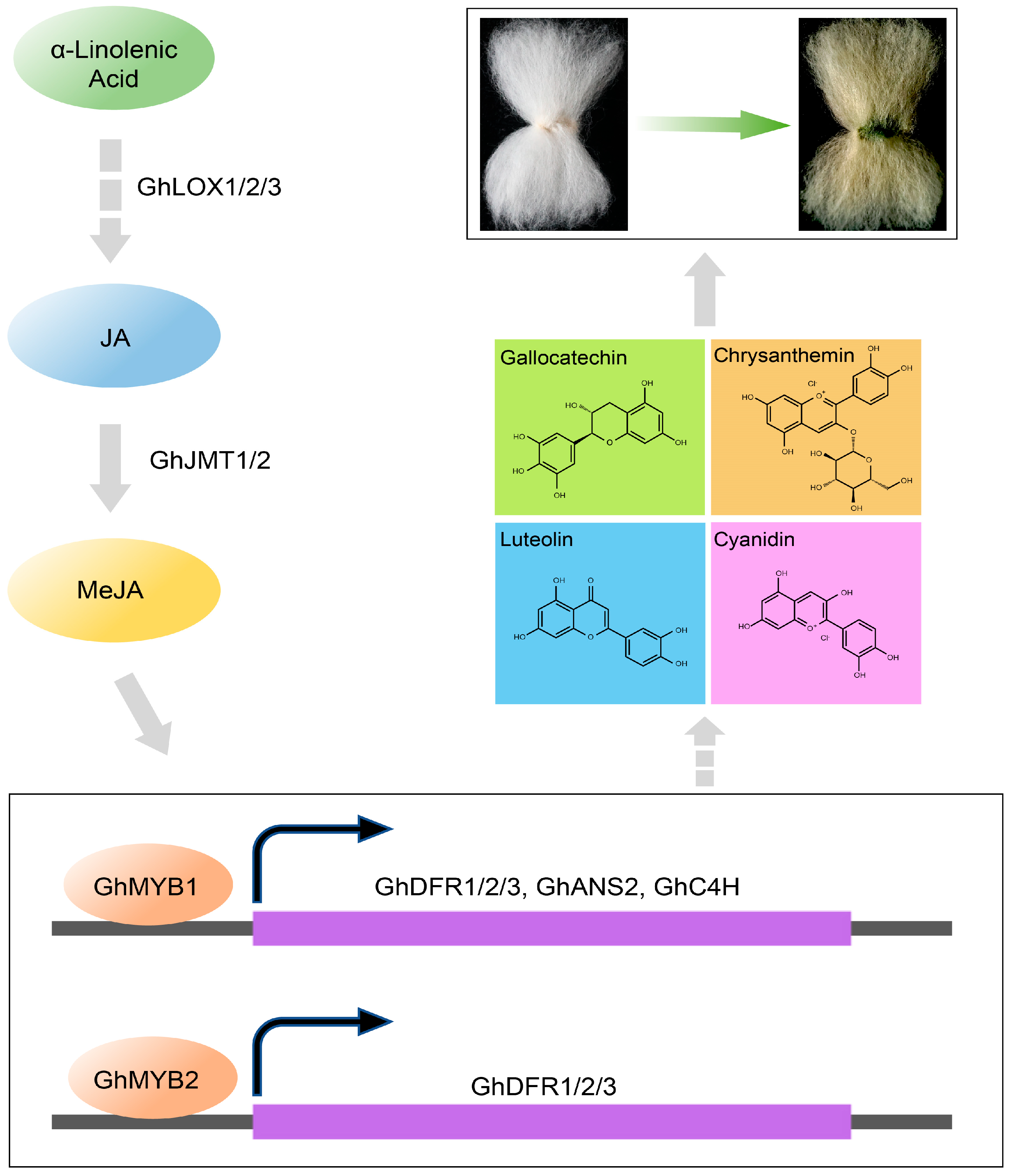
3.2. MeJA Plays a Regulatory Role in Green Cotton Fiber Development and Pigmentation
3.3. Flavonoid Biosynthesis Genes and MYB Factors Synergistically Control Green Pigmentation of Cotton Fibers
3.4. MeJA–MYB–Flavonoid Regulatory Network Promotes the Pigmentation of Green Cotton Fiber
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Transcriptome Sequencing, and Analysis
4.3. Transcription Factors Prediction, KEGG Enrichment, and PPI Network Construction
4.4. Quantitative Reverse Transcription PCR (RT-qPCR) Analysis
4.5. Metabolome Analysis
4.6. HPLC-MS/MS Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NCC | Naturally colored cotton |
| GCFs | Green cotton fibers |
| DPA | Days post anthesis |
| TF | Transcription factor |
| DEGs | Differentially expressed genes |
| UDEGs | Upregulated differentially expressed genes |
| CUDEGs | Co-expression upregulated differentially expressed genes |
| MeJA | Methyl jasmonate |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | Protein–protein interaction |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| VIP | The variable importance in projection |
| PLS-DA | The partial least squares discriminant analysis |
References
- Shi, S.; Tang, R.; Hao, X.; Tang, S.; Chen, W.; Jiang, C.; Long, M.; Chen, K.; Hu, X.; Xie, Q.; et al. Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants 2024, 13, 2028. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Su, M.; Jin, Z.; Zhou, W.; Sun, Y.; Jin, Y.; Shi, Z. Effects of Natural Brown Cotton Bleached Gauze on Wound Healing. Materials 2022, 15, 2070. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.; Hinchliffe, D.J.; Thyssen, G.N. Naturally colored cotton for wearable applications. Front. Plant Sci. 2024, 15, 1350405. [Google Scholar] [CrossRef] [PubMed]
- Günaydin, G.K.; Avinc, O.; Palamutcu, S.; Yavas, A.; Soydan, A.S. Naturally colored organic Cotton and Naturally Colored Cotton Fiber Production. In Organic Cotton: Is it a Sustainable Solution? Springer Nature: Berlin/Heidelberg, Germany, 2019; pp. 81–99. [Google Scholar] [CrossRef]
- Wang, Z.; Rehman, A.; Jia, Y.; Dai, P.; He, S.; Wang, X.; Li, H.; Wang, L.; Qayyum, A.; Peng, Z.; et al. Transcriptome and proteome profiling revealed the key genes and pathways involved in the fiber quality formation in brown cotton. Gene 2023, 868, 147374. [Google Scholar] [CrossRef]
- Yuan, S.N.; Malik, W.; Bibi, N.; Wen, G.J.; Ni, M.; Wang, X.D. Modulation of morphological and biochemical traits using heterosis breeding in colored cotton. J. Agric. Sci. 2013, 151, 57–71. [Google Scholar] [CrossRef]
- Lv, H.W.; Wang, Q.L.; Luo, M.; Zhu, M.D.; Liang, H.M.; Li, W.J.; Cai, H.; Zhou, Z.B.; Wang, H.; Tong, S.Q.; et al. Phytochemistry and pharmacology of natural prenylated flavonoids. Arch. Pharm. Res. 2023, 46, 207–272. [Google Scholar] [CrossRef]
- Ke, L.; Yu, D.; Zheng, H.; Xu, Y.; Wu, Y.; Jiao, J.; Wang, X.; Mei, J.; Cai, F.; Zhao, Y.; et al. Function deficiency of GhOMT1 causes anthocyanidins over-accumulation and diversifies fibre colours in cotton (Gossypium hirsutum). Plant Biotechnol. J. 2022, 20, 1546–1560. [Google Scholar] [CrossRef]
- Liu, H.F.; Luo, C.; Song, W.; Shen, H.; Li, G.; He, Z.G.; Chen, W.G.; Cao, Y.Y.; Huang, F.; Tang, S.W.; et al. Flavonoid biosynthesis controls fiber color in naturally colored cotton. PeerJ. 2018, 6, e4537. [Google Scholar] [CrossRef]
- Canavar, Ö.; Rausher, M.D. Molecular analysis of structural genes involved in flavonoids biosynthesis in naturally colored cotton. Crop Sci. 2021, 61, 1117–1126. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.P.; Zhu, Q.; Li, Y.J.; Sun, J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef]
- Yu, K.; Song, Y.; Lin, J.; Dixon, R.A. The complexities of proanthocyanidin biosynthesis and its regulation in plants. Plant Commun. 2023, 4, 100498. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Wang, X.; Gao, M.; Zhao, X.; Chen, Y.; Zhang, C.; Jiang, H.; Wang, J.; Wang, Y.; Zheng, M.; et al. A glutathione S-transferase GhTT19 determines flower petal pigmentation via regulating anthocyanin accumulation in cotton. Plant Biotechnol. J. 2023, 21, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.U.; Singh, Z.; Shah, H.M.S.; Woodward, A.; Afrifa-Yamoah, E. Methyl jasmonate advances fruit ripening, colour development, and improves antioxidant quality of ‘Yoho’ and ‘Jiro’ persimmon. Food Chem. 2024, 459, 140360. [Google Scholar] [CrossRef]
- Hasan, M.U.; Singh, Z.; Shah, H.M.S.; Kaur, J.; Woodward, A.; Afrifa-Yamoah, E.; Vithana, M.D.K. Preharvest methyl jasmonate application regulates ripening, colour development and improves phytochemical quality of fruits: A review. Sci. Hortic. 2025, 339, 113909. [Google Scholar] [CrossRef]
- Xiao, G.; Zhao, P.; Zhang, Y. A Pivotal Role of Hormones in Regulating Cotton Fiber Development. Front. Plant Sci. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Tu, L.; Hu, H.; Tan, J.; Deng, F.; Tang, W.; Nie, Y.; Zhang, X. GbTCP, a cotton TCP transcription factor, confers fibreelongation and root hair development by a complex regulating system. J. Exp. Bot. 2012, 63, 6267–6281. [Google Scholar] [CrossRef]
- Rudell, D.; Mattheis, J.; Fan, X.; Fellman, J. Methyl Jasmonate enhances anthocyanin accumulation and modifies production of Phenolics and pigments in Fuji’Apples. J. Am. Soc. Hortic. Sci. 2002, 127, 435–441. [Google Scholar] [CrossRef]
- Flores, G.; Blanch, G.P.; del Castillo, M.L.R. Postharvest treatment with (−) and (+)-methyl jasmonate stimulates anthocyanin accumulation in grapes. LWT-Food Sci. Technol. 2015, 62, 807–812. [Google Scholar] [CrossRef]
- Huang, X.; Li, J.; Shang, H.; Meng, X. Effect of methyl jasmonate on the anthocyanin content and antioxidant activity of blueberries during cold storage. J. Sci. Food Agric. 2015, 95, 337–343. [Google Scholar] [CrossRef]
- Pérez, A.G.; Sanz, C.; Olías, R.; Olías, J.M. Effect of methyl jasmonate on in vitro strawberry ripening. J. Agric. Food Chem. 1997, 45, 3733–3737. [Google Scholar] [CrossRef]
- Lu, N.; Roldan, M.; Dixon, R.A. Characterization of two TT2-type MYB transcription factors regulating proanthocyanidin biosynthesis in tetraploid cotton, Gossypium hirsutum. Planta 2017, 246, 323–335. [Google Scholar] [CrossRef]
- Jiang, W.; Jiang, Q.; Shui, Z.; An, P.; Shi, S.; Liu, T.; Zhang, H.; Huang, S.; Jing, B.; Xiao, E.; et al. HaMYBA-HabHLH1 regulatory complex and HaMYBF fine-tune red flower coloration in the corolla of sunflower (Helianthus annuus L.). Plant Sci. 2024, 338, 111901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, L.; Xi, D.; Li, X.; Gao, L.; Miao, L.; Luo, Y.; Tian, M.; Zhu, H. Methyl jasmonate-induced bHLH42 mediates tissue-specific accumulation of anthocyanins via regulating flavonoid metabolism-related pathways in Caitai. Physiol. Plant. 2024, 176, e14434. [Google Scholar] [CrossRef] [PubMed]
- Premathilake, A.T.; Ni, J.; Shen, J.; Bai, S.; Teng, Y. Transcriptome analysis provides new insights into the transcriptional regulation of methyl jasmonate-induced flavonoid biosynthesis in pear calli. BMC Plant Biol. 2020, 20, 388. [Google Scholar] [CrossRef]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, J.Q.; Chen, X.Y.; Zhu, Y.X. Recent Advances and Future Perspectives in Cotton Research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, X.; Tu, L.; Zhu, L.; Guo, X. Isolation by suppression-subtractive hybridization of genes preferentially expressed during early and late fiber development stages in cotton. Mol. Biol. 2006, 40, 741–749. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Guo, X.; Li, X.; Shen, Q.; Fu, X.; Tang, D. Metabolic Patterns of Flavonoid and Its Key Gene Expression Characteristics of Five Cultivars of Tulipa gesneriana during Flower Development. Plants 2024, 13, 459. [Google Scholar] [CrossRef]
- Zhang, X.M.; Liu, S.J.; Gong, W.F.; Sun, J.L.; Pang, B.Y.; Du, X.M. Effects of Plant Growth Regulators on Fiber Growth and Development in Colored Cotton Ovule Culture in vitro. Acta Agron. Sin. 2017, 43, 763–776. [Google Scholar] [CrossRef]
- Geng, D.; Jiang, M.; Dong, H.; Wang, R.; Lu, H.; Liu, W.; Guo, L.; Huang, L.; Xiao, W. MeJA regulates the accumulation of baicalein and other 4′-hydroxyflavones during the hollowed root development in Scutellaria baicalensis. Front. Plant Sci. 2023, 13, 1067847. [Google Scholar] [CrossRef]
- Wen, T.; Wu, M.; Shen, C.; Gao, B.; Zhu, D.; Zhang, X.; You, C.; Lin, Z. Linkage and association mapping reveals the genetic basis of brown fibre (Gossypium hirsutum). Plant Biotechnol. J. 2018, 16, 1654–1666. [Google Scholar] [CrossRef]
- Feng, H.; Li, Y.; Wang, S.; Zhang, L.; Liu, Y.; Xue, F.; Sun, Y.; Wang, Y.; Sun, J. Molecular analysis of proanthocyanidins related to pigmentation in brown cotton fibre (Gossypium hirsutum L.). J. Exp. Bot. 2014, 65, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, Y.; Li, Q.; Zhang, Z.; Ding, H.; Zhang, Y.; Liu, H.; Luo, M.; Liu, D.; Song, W.; et al. Up-regulation of GhTT2-3A in cotton fibres during secondary wall thickening results in brown fibres with improved quality. Plant Biotechnol. J. 2018, 16, 1735–1747. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Burks, D.J.; Azad, R.K. RNA-Seq Data Analysis Pipeline for Plants: Transcriptome Assembly, Alignment, and Differential Expression Analysis. Methods Mol. Biol. 2022, 2396, 47–60. [Google Scholar] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, J.; Huang, Y.; Wang, S.; Wei, L.; Liu, D.; Weng, Y.; Xiang, J.; Zhu, Q.; Yang, Z.; et al. CottonMD: A multi-omics database for cotton biological study. Nucleic Acids Res. 2023, 51, D1446–D1456. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; He, S.; Rehman, A.; Jia, Y.; Li, H.; Pan, Z.; Geng, X.; Gao, Q.; Wang, L.; et al. Transcriptome Co-expression Network and Metabolome Analysis Identifies Key Genes and Regulators of Proanthocyanidins Biosynthesis in Brown Cotton. Front. Plant Sci. 2022, 12, 822198. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant. 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Mishra, P.; Singh, U.; Pandey, C.M.; Mishra, P.; Pandey, G. Application of student’s t-test, analysis of variance, and covariance. Ann. Card. Anaesth. 2019, 22, 407–411. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, S.; Chen, K.; Tang, R.; Li, X.; Wei, Y.; Cheng, Y.; Tang, S.; Chen, W.; Xie, Q.; Meng, Z.; et al. Combined Analysis of Transcriptomes and Metabolomes Reveals That MeJA-Mediated Flavonoid Biosynthesis Is Crucial for Pigment Deposition in Naturally Colored Green Cotton Fibers. Genes 2025, 16, 599. https://doi.org/10.3390/genes16050599
Xie S, Chen K, Tang R, Li X, Wei Y, Cheng Y, Tang S, Chen W, Xie Q, Meng Z, et al. Combined Analysis of Transcriptomes and Metabolomes Reveals That MeJA-Mediated Flavonoid Biosynthesis Is Crucial for Pigment Deposition in Naturally Colored Green Cotton Fibers. Genes. 2025; 16(5):599. https://doi.org/10.3390/genes16050599
Chicago/Turabian StyleXie, Shuangquan, Kailu Chen, Rui Tang, Xuechi Li, Yuxin Wei, Yijie Cheng, Shouwu Tang, Wengang Chen, Quanliang Xie, Zhuang Meng, and et al. 2025. "Combined Analysis of Transcriptomes and Metabolomes Reveals That MeJA-Mediated Flavonoid Biosynthesis Is Crucial for Pigment Deposition in Naturally Colored Green Cotton Fibers" Genes 16, no. 5: 599. https://doi.org/10.3390/genes16050599
APA StyleXie, S., Chen, K., Tang, R., Li, X., Wei, Y., Cheng, Y., Tang, S., Chen, W., Xie, Q., Meng, Z., Ismayil, A., Jin, X., Wang, F., Liu, H., & Li, H. (2025). Combined Analysis of Transcriptomes and Metabolomes Reveals That MeJA-Mediated Flavonoid Biosynthesis Is Crucial for Pigment Deposition in Naturally Colored Green Cotton Fibers. Genes, 16(5), 599. https://doi.org/10.3390/genes16050599