
Toward a Kinh Vietnamese Reference Genome: Constructing a De Novo Genome Assembly Using Long-Read Sequencing and Optical Mapping

 , , , , , , ,
, , , , , , , Highlights
- This is the first study to report the de novo assembly of a Vietnamese (Kinh) genome using PacBio HiFi and Bionano mapping (VHG1.1).
- Fewer variants were detected using VHG1.1 vs. hg38 in Kinh individuals, demonstrating superior population-specific representation.
- An enhanced version, VHG1.2, was created using additional KHV data and showed greater specificity to the Vietnamese genome.
- This study highlights the need for population-specific reference genomes to reduce bias, improve variant detection, and support precision medicine in under-represented groups such the Vietnamese population.
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Declarations
2.2. Donor Selection
2.3. Genomic DNA Preparation
2.4. Pacbio HiFi Library Preparation and Sequencing
2.5. Bionano Optical Map Generation
2.6. Genome Assembly of SMRT Sequencing Reads
2.7. Hybrid Scaffold Assembly
2.8. Assembly Correction and Improvement with Pacbio HiFi Reads
2.9. Variant Calling for VHG
2.10. Polishing Assembly Using Illumina Short-Read Whole-Genome Sequences
2.11. Ordering and Aligning of Super Scaffolds to Chromosomes
2.12. Genome Assembly Evaluation
2.13. Reference Genome Set
2.14. SNP and Indel Calling of KHV Genome
2.15. Structural Variant Calling of KHV Genome
2.16. Comparison VHG1.2 with East Asian Assemblies
2.17. Data Visualization and Summary Statistics
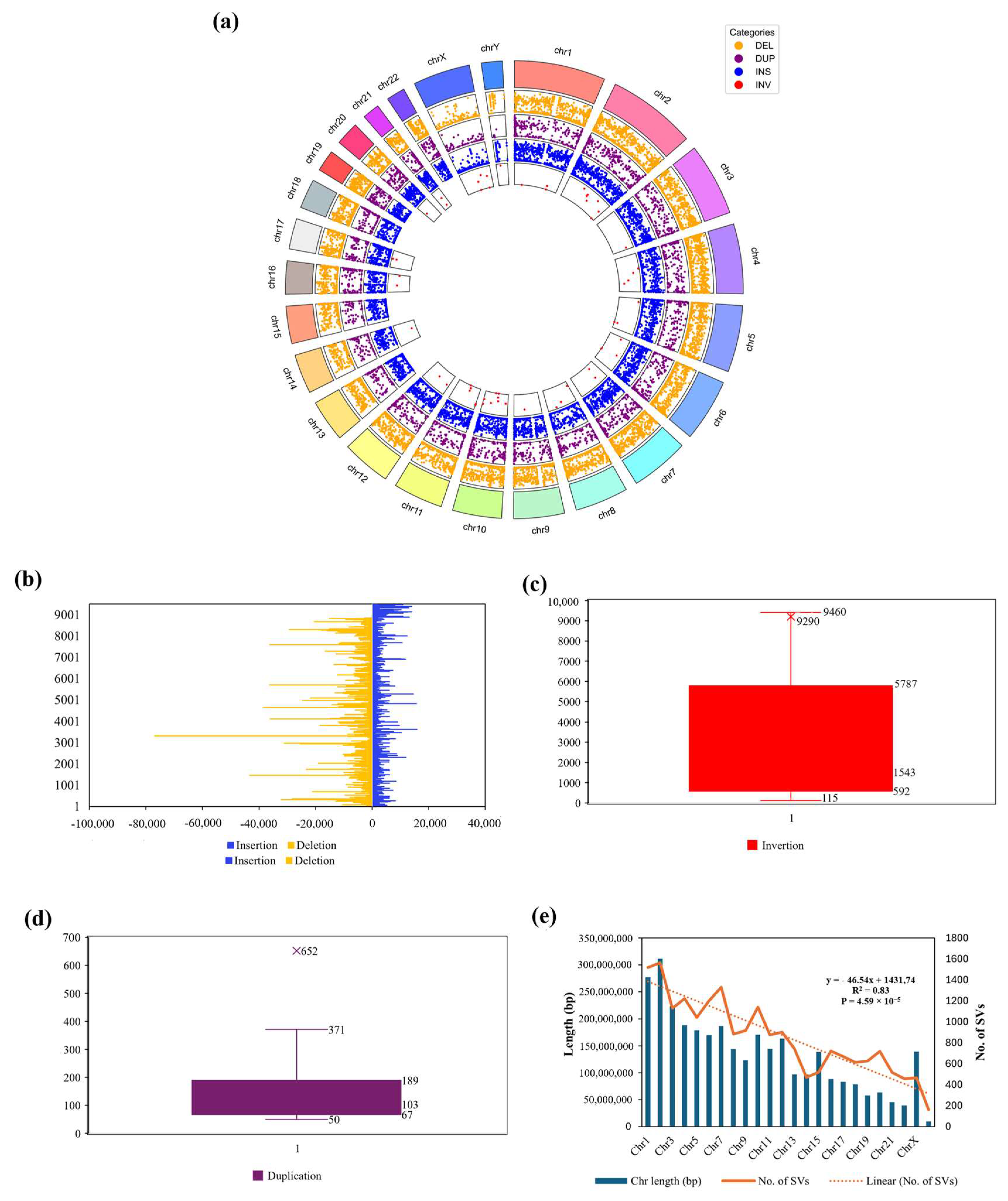
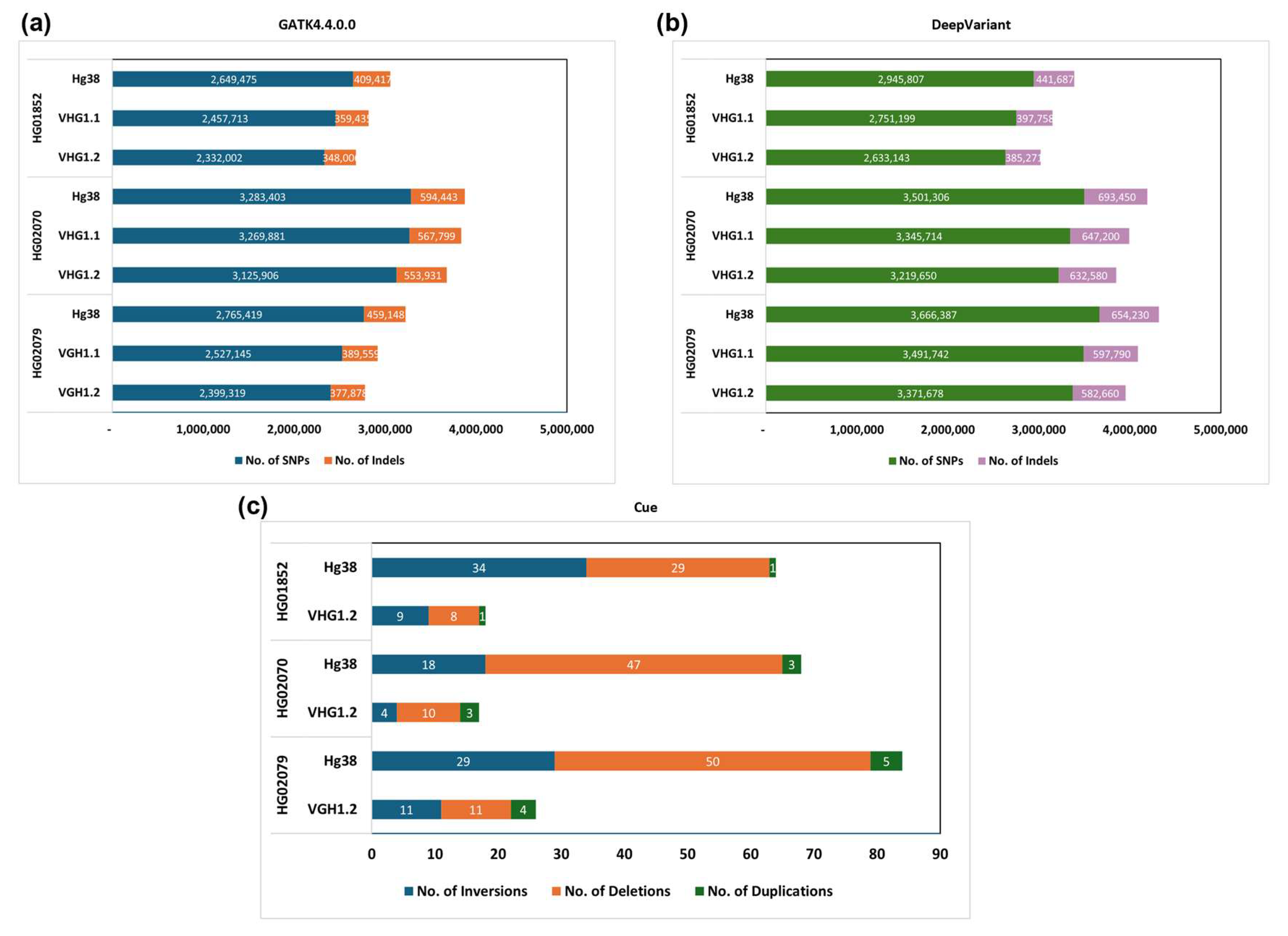
3. Results and Discussion
3.1. De Novo Assembly of a Kinh Vietnamese Genome
3.2. SNPs and Indels Detection of VHG’s HiFi Reads
3.3. Polishing and Quality Evaluation of the De Novo Assembly
3.4. Toward the First Version of the Kinh Vietnamese Population-Specific Reference Genome
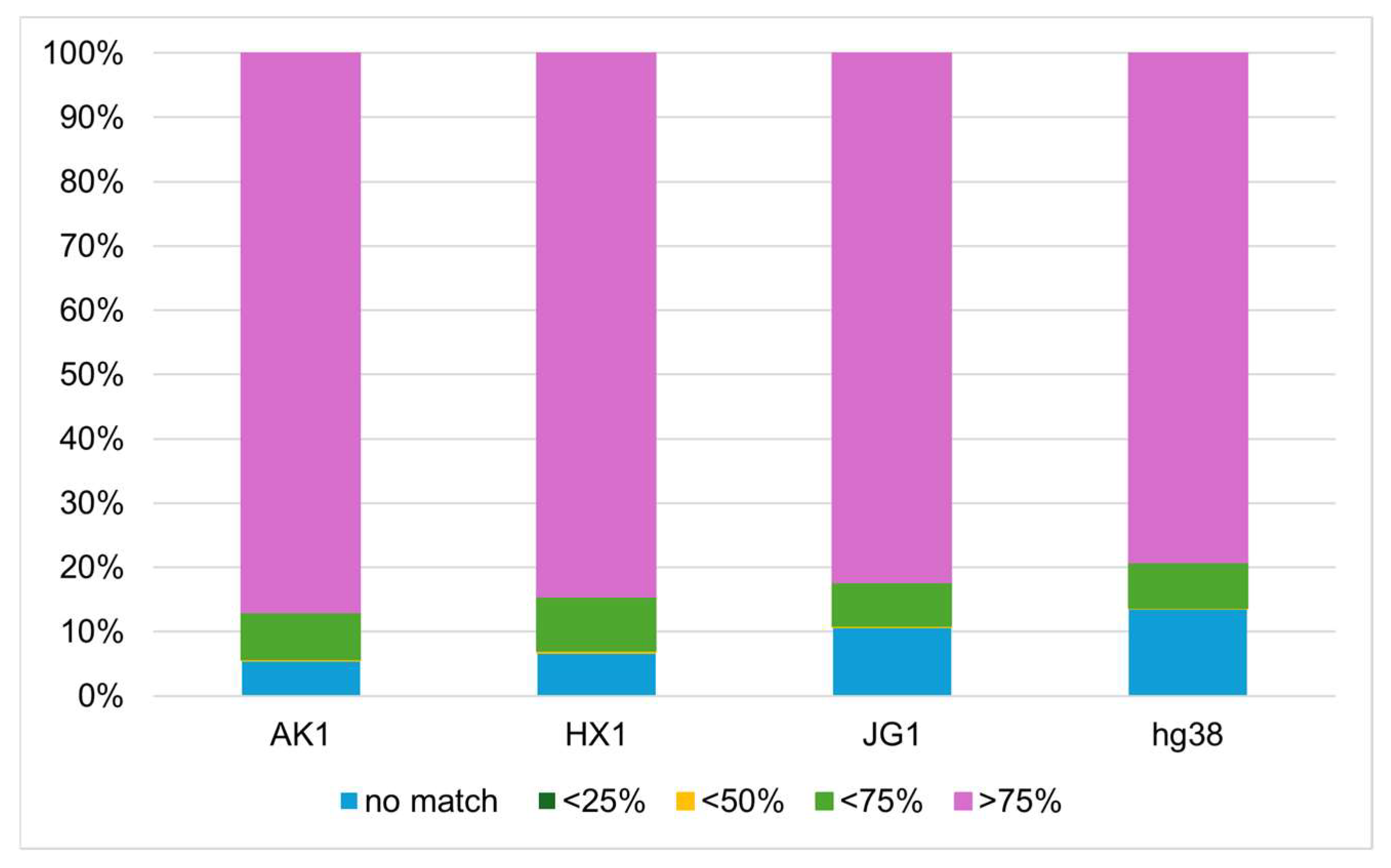
3.5. Representativeness of VHG1.2 for the Vietnamese Population
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WGS | Whole-genome sequences |
| Kbp | Kilobases |
| Mbp | Megabases |
| SNP | Single-nucleotide polymorphism |
| Indel | Short insertions–deletions |
| SV | Structure variant |
| JASPER | Jellyfish-based Assembly Sequence Polisher for Error Reduction |
| KHV | Kinh Vietnamese genome |
| ONT | Oxford Nanopore Technology |
| hg19 | GRCh37 |
| hg38 | GRCh38 |
References
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Jeon, S.; Chung, O.; Jun, J.H.; Kim, H.S.; Blazyte, A.; Lee, H.Y.; Yu, Y.; Cho, Y.S.; Bolser, D.M.; et al. Comparative analysis of 7 short-read sequencing platforms using the Korean Reference Genome: MGI and Illumina sequencing benchmark for whole-genome sequencing. GigaScience 2021, 10, giab014. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Aref-Eshghi, E.; McEldrew, D.A.; Luo, M.; Rajagopalan, R. Long-read sequencing for molecular diagnostics in constitutional genetic disorders. Hum. Mutat. 2022, 43, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- National Research Council. An Assessment of the SBIR Program at the National Institutes of Health; The National Academies Press: Washington, DC, USA, 2009. [Google Scholar] [CrossRef]
- Kingsmore, S.F.; Nofsinger, R.; Ellsworth, K. Rapid genomic sequencing for genetic disease diagnosis and therapy in intensive care units: A review. NPJ Genom. Med. 2024, 9, 17. [Google Scholar] [CrossRef]
- Adams, D.R.; Eng, C.M. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N. Engl. J. Med. 2018, 379, 1353–1362. [Google Scholar] [CrossRef]
- Nagasaki, M.; Kuroki, Y.; Shibata, T.F.; Katsuoka, F.; Mimori, T.; Kawai, Y.; Minegishi, N.; Hozawa, A.; Kuriyama, S.; Suzuki, Y.; et al. Construction of JRG (Japanese reference genome) with single-molecule real-time sequencing. Hum. Genome Var. 2019, 6, 27. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Mak, A.C.Y.; Lai, Y.Y.Y.; Lam, E.T.; Kwok, T.P.; Leung, A.K.Y.; Poon, A.; Mostovoy, Y.; Hastie, A.R.; Stedman, W.; Anantharaman, T.; et al. Genome-wide structural variation detection by genome mapping on nanochannel arrays. Genetics 2016, 202, 351–362. [Google Scholar] [CrossRef]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161. [Google Scholar] [CrossRef] [PubMed]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef]
- Aganezov, S.; Yan, S.M.; Soto, D.C.; Kirsche, M.; Zarate, S.; Avdeyev, P.; Taylor, D.J.; Shafin, K.; Shumate, A.; Xiao, C.; et al. A complete reference genome improves analysis of human genetic variation. Science 2022, 376, eabl3533. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Abasov, R.; Tvorogova, V.E.; Glotov, A.S.; Predeus, A.V. Systematic benchmark of state-of-the-art variant calling pipelines identifies major factors affecting accuracy of coding sequence variant discovery. BMC Genom. 2022, 23, 155. [Google Scholar] [CrossRef]
- Yang, X.; Lee, W.P.; Ye, K.; Lee, C. One reference genome is not enough. Genome Biol. 2019, 20, 104. [Google Scholar] [CrossRef]
- Thorburn, D.J.; Sagonas, K.; Binzer-Panchal, M.; Chain, F.J.J.; Feulner, P.G.D.; Bornberg-Bauer, E.; Reusch, T.B.H.; Samonte-Padilla, I.E.; Milinski, M.; Lenz, T.L.; et al. Origin matters: Using a local reference genome improves measures in population genomics. Mol. Ecol. Resour. 2023, 23, 1706–1723. [Google Scholar] [CrossRef]
- Yang, C.; Zhou, Y.; Song, Y.; Wu, D.; Zeng, Y.; Nie, L.; Liu, P.; Zhang, S.; Chen, G.; Xu, J.; et al. The complete and fully-phased diploid genome of a male Han Chinese. Cell Res. 2023, 33, 745–761. [Google Scholar] [CrossRef]
- Shi, L.; Guo, Y.; Dong, C.; Huddleston, J.; Yang, H.; Han, X.; Fu, A.; Li, Q.; Li, N.; Gong, S.; et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016, 7, 12065. [Google Scholar] [CrossRef]
- Du, Z.; Ma, L.; Qu, H.; Chen, W.; Zhang, B.; Lu, X.; Zhai, W.; Sheng, X.; Sun, Y.; Li, W.; et al. Whole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han Genome. Genom. Proteom. Bioinform. 2019, 17, 229–247. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, X.; Qu, S.; Jia, P.; Wang, B.; Gao, S.; Xu, T.; Zhang, W.; Huang, J.; Ye, K. Haplotype-resolved Chinese male genome assembly based on high-fidelity sequencing. Fundam. Res. 2022, 2, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Kim, H.; Kim, H.M.; Jho, S.; Jun, J.; Lee, Y.J.; Chae, K.S.; Kim, C.G.; Kim, S.; Eriksson, A.; et al. An ethnically relevant consensus Korean reference genome is a step towards personal reference genomes. Nat. Commun. 2016, 7, 13637. [Google Scholar] [CrossRef]
- Seo, J.S.; Rhie, A.; Kim, J.; Lee, S.; Sohn, M.H.; Kim, C.U.; Hastie, A.; Cao, H.; Yun, J.Y.; Kim, J.; et al. De novo assembly and phasing of a Korean human genome. Nature 2016, 538, 243–247. [Google Scholar] [CrossRef]
- Maretty, L.; Jensen, J.M.; Petersen, B.; Sibbesen, J.A.; Liu, S.; Villesen, P.; Skov, L.; Belling, K.; Theil Have, C.; Izarzugaza, J.M.G.; et al. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 2017, 548, 87–91. [Google Scholar] [CrossRef]
- Ameur, A.; Che, H.; Martin, M.; Bunikis, I.; Dahlberg, J.; Höijer, I.; Häggqvist, S.; Vezzi, F.; Nordlund, J.; Olason, P.; et al. De novo assembly of two Swedish genomes reveals missing segments from the human GRCh38 reference and improves variant calling of population-scale sequencing data. Genes 2018, 9, 486. [Google Scholar] [CrossRef]
- Takayama, J. Technical Notes on the Construction of the Japanese Near T2T Reference Genome, JG3. Japanese Multi-Omics Reference Panel Portal. 2024. Available online: https://jmorp.megabank.tohoku.ac.jp/downloads/tommo-jg3.0.0-20240618 (accessed on 10 June 2024).
- Kim, H.S.; Jeon, S.; Kim, C.; Kim, Y.K.; Cho, Y.S.; Kim, J.; Blazyte, A.; Manica, A.; Lee, S.; Bhak, J. Chromosome-scale assembly comparison of the Korean Reference Genome KOREF from PromethION and PacBio with Hi-C mapping information. GigaScience 2019, 8, giz125. [Google Scholar] [CrossRef]
- Chao, K.H.; Zimin, A.V.; Pertea, M.; Salzberg, S.L. The first gapless, reference-quality, fully annotated genome from a Southern Han Chinese individual. G3 Genes Genomes Genet. 2023, 13, jkac321. [Google Scholar] [CrossRef]
- Sriwichaiin, S.; Makino, S.; Funayama, T.; Otsuki, A.; Kawashima, J.; Okamura, Y.; Tadaka, S.; The Tohoku Medical Megabank Project Study Group; Katsuoka, F.; Kumada, K.; et al. JG2: An updated version of the Japanese population-specific reference genome. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hwang, M.Y.; Choi, N.H.; Won, H.H.; Kim, B.J.; Kim, Y.J. Analyzing the Korean reference genome with meta-imputation increased the imputation accuracy and spectrum of rare variants in the Korean population. Front. Genet. 2022, 13, 1008646. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Lin, B.; Hui, J.; Mao, H. Nanopore technology and its applications in gene sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef]
- Yuan, Y.; Chung, C.Y.; Chan, T.F. Advances in optical mapping for genomic research. Comput. Struct. Biotechnol. J. 2020, 18, 2051–2062. [Google Scholar] [CrossRef]
- Takayama, J.; Tadaka, S.; Yano, K.; Katsuoka, F.; Gocho, C.; Funayama, T.; Makino, S.; Okamura, Y.; Kikuchi, A.; Sugimoto, S.; et al. Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. Nat. Commun. 2021, 12, 226. [Google Scholar] [CrossRef]
- ten Berk de Boer, E.; Ameur, A.; Bunikis, I.; Ek, M.; Stattin, E.-L.; Feuk, L.; Eisfeldt, J.; Lindstrand, A. Long-read sequencing and optical mapping generates near T2T assemblies that resolves a centromeric translocation. Sci. Rep. 2024, 14, 9000. [Google Scholar] [CrossRef]
- Pischedda, S.; Barral-Arca, R.; Gómez-Carballa, A.; Pardo-Seco, J.; Catelli, M.L.; Álvarez-Iglesias, V.; Cárdenas, J.M.; Nguyen, N.D.; Ha, H.H.; Le, A.T.; et al. Phylogeographic and genome-wide investigations of Vietnam ethnic groups reveal signatures of complex historical demographic movements. Sci. Rep. 2017, 7, 12630. [Google Scholar] [CrossRef]
- Hai, D.T.; Thanh, N.D.; Trang, P.T.M.; Quang, L.S.; Hang, P.T.T.; Cuong, D.C.; Phuc, H.K.; Duc, N.H.; Dong, D.D.; Minh, B.Q.; et al. Whole genome analysis of a Vietnamese trio. J. Biosci. 2015, 40, 113–124. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Shimizu, K.K. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinform. 2017, 18, 474. [Google Scholar] [CrossRef]
- Kim, J.I.; Ju, Y.S.; Park, H.; Kim, S.; Lee, S.; Yi, J.H.; Mudge, J.; Miller, N.A.; Hong, D.; Bell, C.J.; et al. A highly annotated whole-genome sequence of a Korean individual. Nature 2009, 460, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with HiFiasm. Nat. Methods 2021, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Fungtammasan, A.; Carroll, A.; Chou, M.; Schmitt, A.; Zhou, X.; Mac, S.; Peluso, P.; Hatas, E.; Ghurye, J.; et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 2021, 39, 309–312. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lou, H.; Cui, C.; Deng, L.; Gao, Y.; Zheng, W.; Guo, Y.; Wang, X.; Ning, Z.; Li, J.; et al. De novo assembly of a Tibetan genome and identification of novel structural variants associated with high-altitude adaptation. Natl. Sci. Rev. 2020, 7, 391–402. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Wang, A.Y.; Gao, M.; Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biol. 2021, 22, 312. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. 2007. Available online: https://api.semanticscholar.org/CorpusID:18002845 (accessed on 14 June 2024).
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Xie, H.; Li, W.; Hu, Y.; Yang, C.; Lu, J.; Guo, Y.; Wen, L.; Tang, F. De novo assembly of human genome at single-cell levels. Nucleic Acids Res. 2022, 50, 7479–7492. [Google Scholar] [CrossRef]
- Zheng-Bradley, X.; Streeter, I.; Fairley, S.; Richardson, D.; Clarke, L.; Flicek, P. Alignment of 1000 Genomes Project reads to reference assembly GRCh38. GigaScience 2017, 6, gix038. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Broad Institute. GATK Documentation. 2024. Available online: https://gatk.broadinstitute.org/hc/en-us/community/posts/26880110839707-gatk-CombineGVCFs-output-contains-only-one-Chr (accessed on 1 March 2025).
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Popic, V.; Rohlicek, C.; Cunial, F.; Hajirasouliha, I.; Meleshko, D.; Garimella, K.; Maheshwari, A. Cue: A deep-learning framework for structural variant discovery and genotyping. Nat. Methods 2023, 20, 559–568. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Vu-Trieu, A.; Djoulah, S.; Tran-Thi, C.; Ngyuyen-Thanh, T.; Le Monnier De Gouville, I.; Hors, J.; Sanchez-Mazas, A. HLA-DR and –DQB1 DNA polymorphisms in a Vietnamese Kinh population from Hanoi. Eur. J. Immunogenet. 1997, 24, 345–356. [Google Scholar] [CrossRef]
- Betschart, R.O.; Thiéry, A.; Aguilera-Garcia, D.; Zoche, M.; Moch, H.; Twerenbold, R.; Zeller, T.; Blankenberg, S.; Ziegler, A. Comparison of calling pipelines for whole genome sequencing: An empirical study demonstrating the importance of mapping and alignment. Sci. Rep. 2022, 12, 21502. [Google Scholar] [CrossRef]
- Lin, Y.L.; Chang, P.C.; Hsu, C.; Hung, M.Z.; Chien, Y.H.; Hwu, W.L.; Lai, F.; Lee, N.C. Comparison of GATK and DeepVariant by trio sequencing. Sci. Rep. 2022, 12, 1809. [Google Scholar] [CrossRef]
- Chen, N.C.; Kolesnikov, A.; Goel, S.; Yun, T.; Chang, P.C.; Carroll, A. Improving variant calling using population data and deep learning. BMC Bioinform. 2023, 24, 197. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, X.; Weng, Z.; Sidow, A. De novo diploid genome assembly for genome-wide structural variant detection. NAR Genom. Bioinform. 2019, 2, lqz018. [Google Scholar] [CrossRef]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Z.; Li, T.; Xie, C.; Zhao, L.; Yang, J.; Ouyang, S.; Liu, Y.; Li, T.; Xie, Z. Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation. Nat. Commun. 2021, 12, 6501. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Population | Sex | Total Length (Gbp) | Contigs | Hybrid Scaffold | Number of Gaps | N-Gap Length (Mbp) | Year | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | N50 (Mbp) | Number | N50 (Mbp) | ||||||||
| VHG1.2 | Kinh Vietnamese | M | 3.22 | 21981 | 8.781 | 295 | 50.6 | - | - | 2023 | This study |
| JG1 | Japanese | M | 3.09 | 1068 | 23.6 | 624 | 142 | 473 | 2.51 | 2021 | [39] |
| ZF1 | Tibetan | M | 2.9 | 3148 1 | 23.6 1 | 2321 2 | 47.2 2 | 740 | 7.82 | 2020 | [47] |
| NH1.0 | Northern Han Chinese | M | 2.89 | 6663 1 | 1.74 1 | 5574 2 | 46.6 2 | 8484 2 | - | 2019 | [22] |
| Swe1 | Swedish | M | 3.13 | 3139 3 | 9.47 3 | n/a | 49.8 | - | - | 2018 | [28] |
| Swe2 | Swedish | F | 3.1 | 3162 3 | 8.52 3 | n/a | 45.4 | - | - | 2018 | [28] |
| AK1 | Korean | M | 2.9 | 4206 | 17.92 | 2832 | 44.9 | 264 | 3.74 | 2016 | [26] |
| HX1 | Chinese | M | 2.93 | 5843 | 8.33 | n/a | 22.0 | 10,901 | 39.34 | 2016 | [21] |
| HG001/NA12878 | European | F | 3.18 | - | 1.4 | 202 | 31.3 | 2332 | 146.4 | 2015 | [38] |
| GRCh38.p14 | Reference | M | 3.21 | 996 | 57.9 | 470 | 67.8 | 349 | 150.6 | 2022 | NCBI |
| T2T-CHM13v2.0 | Reference | - | 3.05 | 23 | 154.3 | 23 | 154.3 | 0 | 0 | 2022 | [59] |
| Parameters | Super Scaffolds | VHG1.1 | VHG1.2 |
|---|---|---|---|
| Number of scaffolds | 295 | 295 | 295 |
| Total length | 3,225,940,482 | 3,222,774,051 | 3,222,784,990 |
| N50 | 50,640,186 | 50,627,973 | 50,628,337 |
| GC (%) | 40.85 | 40.85 | 40.85 |
| Largest contig | 145,919,952 | 145,820,377 | 145,820,867 |
| >50% identity with hg38 | 90.58% | 90.65% | 86.26% |
| >75% identity with hg38 | 82.63% | 83.93% | 79.25% |
| >50% identity with T2T | 90.19% | 90.26% | 92.06% |
| >75% identity with T2T | 83.69% | 82.90% | 85.88% |
| BUSCO score (%) | 91.5 | 92 | 92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dung, L.T.; Lam, L.T.; Trang, N.H.; Anh, N.V.H.; Nam, N.N.; Nhung, D.T.; Linh, T.H.; Giang, L.N.; Ha, H.; Huy, N.Q.; et al. Toward a Kinh Vietnamese Reference Genome: Constructing a De Novo Genome Assembly Using Long-Read Sequencing and Optical Mapping. Genes 2025, 16, 536. https://doi.org/10.3390/genes16050536
Dung LT, Lam LT, Trang NH, Anh NVH, Nam NN, Nhung DT, Linh TH, Giang LN, Ha H, Huy NQ, et al. Toward a Kinh Vietnamese Reference Genome: Constructing a De Novo Genome Assembly Using Long-Read Sequencing and Optical Mapping. Genes. 2025; 16(5):536. https://doi.org/10.3390/genes16050536
Chicago/Turabian StyleDung, Le Thi, Le Tung Lam, Nguyen Hong Trang, Nguyen Vu Hung Anh, Nguyen Ngoc Nam, Doan Thi Nhung, Tran Huyen Linh, Le Ngoc Giang, Hoang Ha, Nguyen Quang Huy, and et al. 2025. "Toward a Kinh Vietnamese Reference Genome: Constructing a De Novo Genome Assembly Using Long-Read Sequencing and Optical Mapping" Genes 16, no. 5: 536. https://doi.org/10.3390/genes16050536
APA StyleDung, L. T., Lam, L. T., Trang, N. H., Anh, N. V. H., Nam, N. N., Nhung, D. T., Linh, T. H., Giang, L. N., Ha, H., Huy, N. Q., & Hai, T. N. (2025). Toward a Kinh Vietnamese Reference Genome: Constructing a De Novo Genome Assembly Using Long-Read Sequencing and Optical Mapping. Genes, 16(5), 536. https://doi.org/10.3390/genes16050536