
The Metabolic Consequences of Pathogenic Variant in FXYD2 Gene Encoding the Gamma Subunit of Sodium/Potassium-Transporting ATPase in Two Siblings with Sodium-Dependent Defect of Fructose, Galactose and Glucose Renal Reabsorption


Abstract
1. Introduction
- Evaluating 24-h urinary excretion of calcium, citrate, protein, albumin, fructose, galactose, and glucose.
- Assessing the impact of gradually increasing volemia during a water-loading test on renal excretion of calcium, phosphate, magnesium, uric acid, citrate, glucose, and protein.
- Investigating the effect of gradually increasing volemia in a water-loading test on renal excretion of calcium, citrate, endothelin-1 (ET-1), and protein in two sisters, with comparison to a control group.
- Assessing urine acidification ability and tubular citrate transport in a shortened ammonium chloride (NH4Cl) loading test.
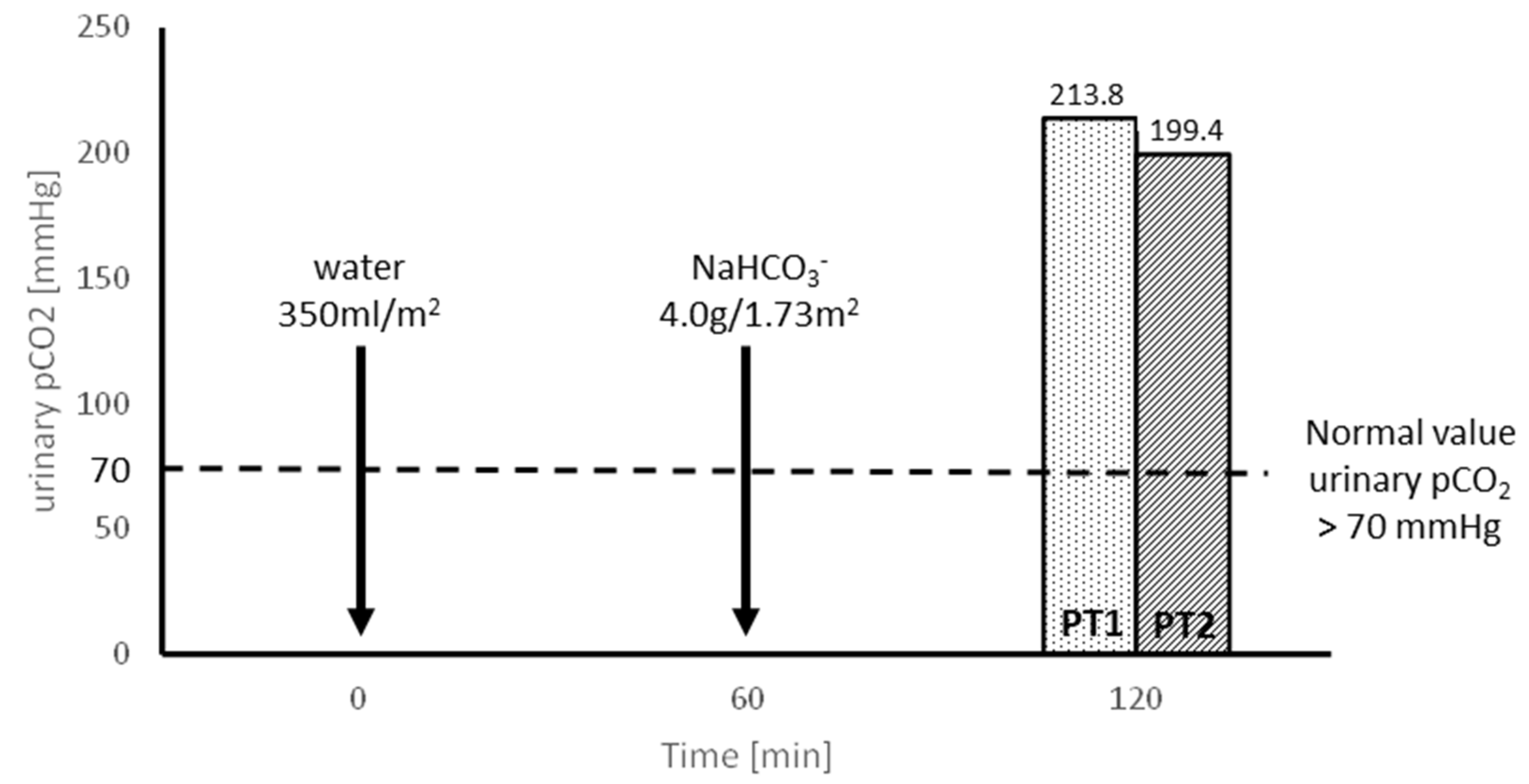
- Evaluating the tubular capacity for H+ secretion in an oral sodium bicarbonate (NaHCO3) loading test, preceded by the oral administration of 300 mL/m2 of water.
- Identifying the genetic cause of the observed unusual metabolic consequences.
2. Material and Methods
2.1. Patients
2.2. Control Group
2.3. Biochemical Analysis
2.4. Genetic Analysis
3. Results
3.1. Results of Functional Tests

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Patient 1 | Patient 2 | Patients Mother |
|---|---|---|---|
| Calcium mg/kg/24 h * mg/24 h | 12.1 | 9.6 | 211.7 x |
| Protein mg/24 h | 1771 | 2392 | 149 |
| Microalbuminuria mg/24 h | 6.3 | 3.4 | 8.6 |
| Citrate mg/g creat ** mg/kg/24 h *** mg/24 h | 967.7 18.3 | 907.1 18.3 | 299.5 xx |
| Glucose mg/24 h | 1230.0 | 2040.0 | 26 |
| Galactose mg/24 h | 144.8 | 134.4 | 40 |
| Fructose mg/24 h | 5765.0 | 3979.5 | 715.1 |
| Parameter | Patient 1 | Patient 2 | ||||
|---|---|---|---|---|---|---|
| Before Loading | After Loading | Before Loading | After Loading | |||
| 300 mL/m2 | 500 mL/m2 | 300 mL/m2 | 500 mL/m2 | |||
| Serum | ||||||
| Creatinine µmol/L | 53.0 | 44.2 | 53.0 | 53.0 | ||
| Uric acid µmol/L | 261.8 | 261.8 | 202.3 | 202.3 | ||
| Na mmol/L | 137 | 135 | 137 | 133 | ||
| Ca mmol/L | 2.39 | 2.37 | 2.28 | 2.40 | ||
| P mmol/L | 1.57 | 1.56 | 1.60 | 1.65 | ||
| Mg mmol/L | 0.93 | 0.88 | 0.86 | 0.82 | ||
| Citrate mmol/L | 0.15 | 0.19 | 0.07 | 0.11 | ||
| Urine | ||||||
| Volume mL/min | 0.52 | 3.83 | 4.10 | 0.57 | 4.50 | 6.00 |
| Osmolality mOsm/kg | 461 | 130 | 173 | 418 | 201 | 257 |
| Ca/creat mg/mg | 0.45 | 2.19 | 5.10 | 0.31 | 2.73 | 5.38 |
| Mg/creat mg/mg | 0.08 | 0.11 | 0.19 | 0.07 | 0.09 | 0.12 |
| FE Mg % | 2.0 | 3.1 | 4.5 | 2.1 | 2.7 | 3.4 |
| P/creat mg/mg | 0.31 | 0.30 | 0.40 | 0.48 | 0.49 | 0.39 |
| TRP% | 96.0 | 96.3 | 95.9 | 94.2 | 94.1 | 95.4 |
| TmP/GFR mmol/L | 1.51 | 1.50 | 1.50 | 1.51 | 1.53 | 1.58 |
| FE uric acid % | 4.3 | 6.4 | 6.1 | 6.8 | 7.7 | 8.0 |
| Citrate/creat mg/g | 1012.3 | 2950.6 | 6061.9 | 682.3 | 3099.0 | 5806.6 |
| FE citrate% | 21.3 | 58.2 | 106.3 | 30.8 | 108.7 | 166.3 |
| Glucose/creat mg/mg | 6.8 | 36.2 | 78.7 | 4.5 | 43.3 | 87.5 |
| Protein/creat mg/mg | 0 | 6.47 | 11.1 | 0.93 | 3.88 | 4.81 |
| FE Na% | 0.44 | 0.67 | 0.70 | 0.97 | 1.04 | 0.60 |
| Parameter | Patient 1 | Patient 2 | ||||
|---|---|---|---|---|---|---|
| Before Loading | After Loading | Before Loading | After Loading | |||
| 4 h | 5 h | 4 h | 5 h | |||
| Serum | ||||||
| Creatinine µmol/L | 44.2 | 35.4 | ||||
| Citrate mmol/L | 0.15 | 0.13 | 0.12 | 0.11 | ||
| Plasma HCO3 mmol/L | 24.6 | 19.4 | 20.0 | 24.7 | 19.6 | 21.3 |
| Urine | ||||||
| pH | 7.12 | 5.02 | 4.49 | 6.99 | 4.74 | 4.56 |
| TA μEq/min/1.73 m2 | 9.8 | 15.8 | 33.1 | 16.7 | 18.2 | 32.8 |
| NH4 μEq/min/1.73 m2 | 8.7 | 40.5 | 43.4 | 22.5 | 53.6 | 47.2 |
| HCO3 μEq/min/1.73 m2 | 16.8 | 29.3 | 54.6 | 52.4 | 19.1 | 68.4 |
| Net H+ μEq/min/1.73 m2 | −1.7 | 27.0 | 21.9 | −13.2 | 52.7 | 11.6 |
| Citrate/Creatinine mg/g | 1202.0 | 704.8 | 3057.0 | 625.8 | 347.3 | 2117.9 |
| FE citrate% | 21.1 | 14.3 | 62.2 | 10.8 | 6.6 | 40.1 |
| FE HCO3% | 0.6 | 1.6 | 2.1 | 1.5 | 0.8 | 2.5 |
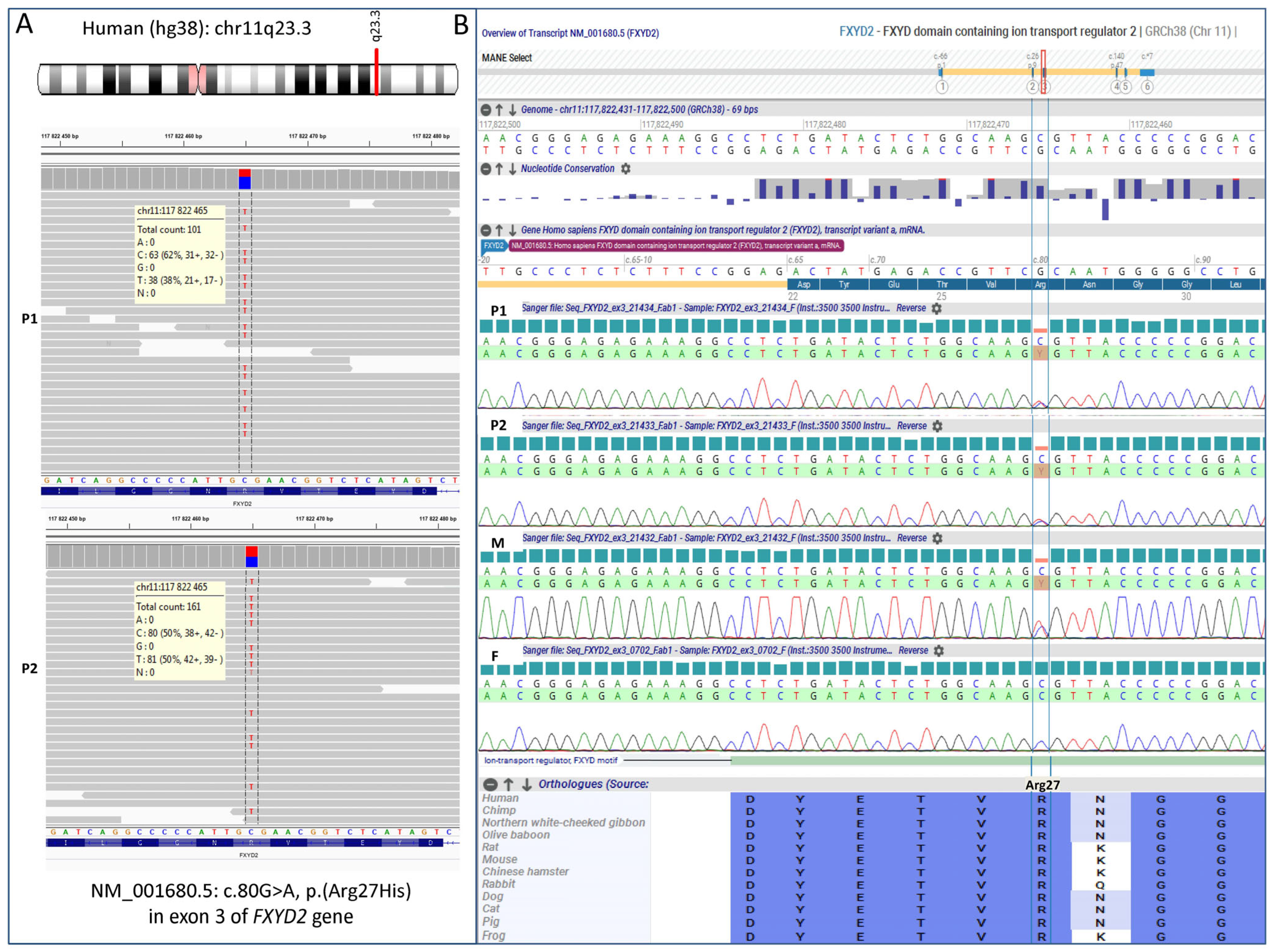
3.2. Molecular Results
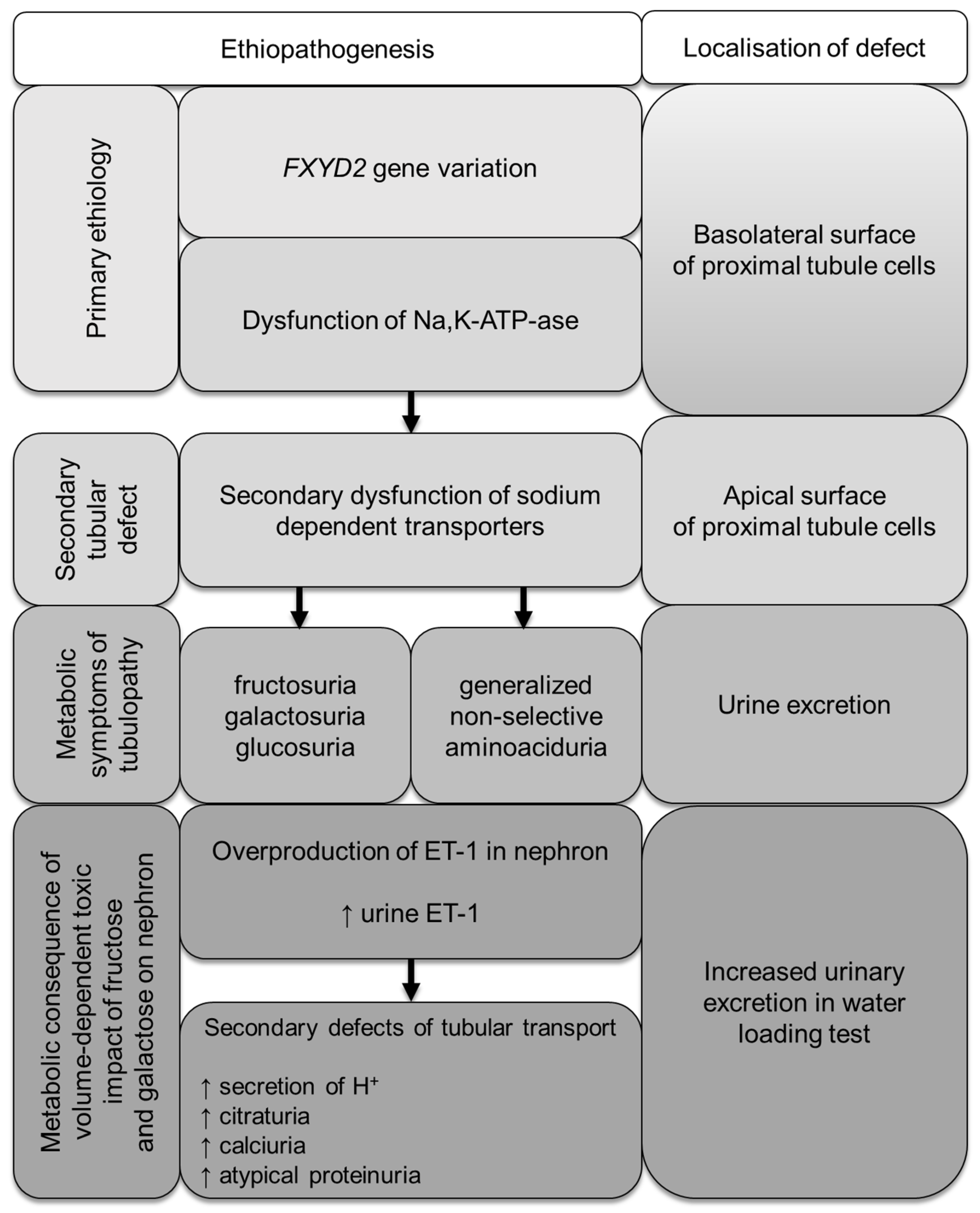
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Vincente, A.; Cabral, P.; Hong, N.; Asirwatham, J.; Saez, F.; Garvin, J. Fructose reabsorption by rat proximal tubules of Na+-linked cotransporters and the effect of dietary glucose. Am. J. Physiol. Ren. Physiol. 2019, 316, F473–F480. [Google Scholar] [CrossRef] [PubMed]
- Feraille, E.; Doucet, A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney hormnal control. Physiol. Rev. 2001, 81, 345–418. [Google Scholar] [CrossRef] [PubMed]
- Sugawara-Yokoo, M.; Suzuki, Y.; Matsuzaki, T.; Naruse, T.; Takata, K. Presence of fructose transporter GLUT5 in the S3 proximal tubules in the rat kidney. Kidney Int. 1999, 56, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef]
- Arystarkhova, E.; Wetzel, R.K.; Sweadner, K.J. Distribution and oligomeric association of splice forms of Na(+)-K(+)-ATPase regulatory γ-subunit in rat kidney. Am. J. Physiol. Ren. Physiol. 2002, 282, F393–F407. [Google Scholar] [CrossRef] [PubMed]
- Arystarkhova, E.; Donnet, C.; Asinovski, N.K.; Sweadner, K.J. Differential regulation of renal Na,K-ATPase by splice variants of the γ subunit. J. Biol. Chem. 2002, 277, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Isshiki, K.; Kume, S.; Chin-Kanasaki, M.; Araki, H.; ArakiSI Koya, D.; Haneda, M.; Kashiwagi, A.; Maegawa, H.; Uzu, T. Fructose induces tubulointerstitial injury in the kidney of mice. Biochem. Biophys. Res. Commun. 2012, 419, 244–249. [Google Scholar] [CrossRef]
- Marsen, T.A.; Schramek, H.; Dunn, M.J. Renal actions of endothelin: Linking cellular signaling pathways to kidney disease. Kidney Int. 1994, 45, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Grenda, R.; Wühl, E.; Litwin, M.; Janas, R.; Sladowska, J.; Arbeiter, K.; Berg, U.; Caldas-Afonso, A.; Fischbach, M.; Mehls, O.; et al. Urinary excretion of endothelin-1 (ET-1), transforming growth factor-beta1 (TGF-beta1) and vascular endothelial growth factor (VEGF165) in paediatric chronic kidney diseases: Results of the ESCAPE trial. Dial. Transplant. 2007, 22, 3487–3494. [Google Scholar] [CrossRef]
- Zawadzki, J. Permeability defect with bicarbonate leak as a mechanism of immune-related distal renal tubular acidosis. Am. J. Kidney Dis. 1998, 31, 527–532. [Google Scholar] [CrossRef]
- Roe, H.G. A colorimetric methods for the determination of fructose in blood and urine. J. Biol. Chem. 1934, 107, 15–22. [Google Scholar] [CrossRef]
- Kizhner, T.; Werman, M. Long-term fructose intake: Biochemical consequences and altered renal histology in the male rat. Metabolism 2002, 51, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Halat-Wolska, P.; Ciara, E.; Pac, M.; Obrycki, Ł.; Wicher, D.; Iwanicka-Pronicka, K.; Bielska, E.; Chałupczyńska, B.; Siestrzykowska, D.; Kostrzewa, G.; et al. Molecular Review of Suspected Alport Syndrome Patients-A Single-Centre Experience. Genes 2025, 16, 196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Ordóñez, F.A.; Claramunt-Taberner, D.; Gil-Peña, H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr. Nephrol. 2015, 30, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Alon, U.S. Pathophysiology of hypercalciuria in children. Pediatr. Nephrol. 2007, 22, 1659–1673. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Baaij, J.H.; Dorresteijn, E.M.; Hennekam, E.A.; Kamsteeg, E.J.; Meijer, R.; Dahan, K.; Muller, M.; van den Dorpel, M.A.; Bindels, R.J.; Hoenderop, J.G.; et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol. Dial. Transplant. 2015, 30, 952–957. [Google Scholar] [CrossRef]
- Cairo, E.R.; Friedrich, T.; Swarts, H.G.; Knoers, N.V.; Bindels, R.J.; Monnens, L.A.; Willems, P.H.; De Pont, J.J.; Koenderink, J.B. Impaired routing of wild type FXYD2 after oligomerisation with FXYD2-G41R might explain the dominant nature of renal hypomagnesemia. Biochim. Biophys. Acta 2008, 1778, 398–404. [Google Scholar] [CrossRef]
- Arystarkhova, E.; Sweadner, K.J. Functional Studies of Na(+),K(+)-ATPase Using Transfected Cell Cultures. Methods Mol. Biol. 2016, 1377, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.M.; Ding, Y.; Yu, J.; Yao, Y.; Marassi, F.M. Structure of the Na,K-ATPase regulatory protein FXYD2b in micelles: Implications for membrane-water interfacial arginines. Biochim. Biophys. Acta 2015, 1848, 299–306. [Google Scholar] [CrossRef]
- Grempler, R.; Augustin, R.; Froehner, S.; Hildebrandt TSimon, E.; Mark, M.; Eickelmann, P. Functional characterization of human SGLT-5 as a novel kidney-specific sodium-dependent sugar transport. FEBS Lett. 2012, 586, 248–253. [Google Scholar] [CrossRef]
- Chen, S.; Evans, T.; Deng, D.; Cukiernik, M.; Chakrabarti, S. Hyperhexosemia induced functional and structural changes in the kidneys: Role of endothelins. Nephron 2002, 90, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Gastone, G.; Serneri, N.; Modesti, P.; Cecioni, I.; Biagini, D.; Costoli, A.; Colella, A.; Naldoni, A.; Paoletti, P. Plasma endothelin and renal endothelin are two distinct systems involved in volume homeostasis. Am. J. Physiol. Heart Circ. Physiol. 1995, 268, H1829–H1837. [Google Scholar] [CrossRef]
- Matthyus, I.; Zimmerhackl, L.; Schwartz, A.; Hentschel, M.; Brandis, M.; Miltenyi, M.; Tulassay, T. Renal excretion of endothelin in children is influenced by age and diuresis. Acta Paediatr. 1994, 83, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.; Padilla, E. Osmolar regulation of endothelin-1 production by rat inner medullary collecting duct. J. Clin. Investig. 1993, 91, 1235–1240. [Google Scholar] [CrossRef]
- Pandit, M.; Inscho, E.; Zhang, S.; Seki, T.; Rohatgi, R.; Gusella, L.; Kishore, B.; Kohan, D. Flow regulation of endothelin-1 production in the inner medullary collecting duct. Am. J. Physiol. Ren. Physiol. 2015, 308, F541–F552. [Google Scholar] [CrossRef]
- Wesson, D. Endogenous endothelins mediate increased distal tubule acidification induced by dietary acid in rats. J. Am. Soc. Clin. Investig. 1997, 99, 2203–2211. [Google Scholar] [CrossRef]
- Nakagawa, T.; Johnson, R.J.; Andres-Hernando, A.; Roncal-Jimenez, C.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Fructose production and metabolism in the kidney. J. Am. Soc. Nephrol. 2020, 31, 898–906. [Google Scholar] [CrossRef]
- Viering, D.H.H.M.; de Baaij, J.H.F.; Walsh, S.P.; Kleta, R. Genetic causes of hypomagnesemia, a clinical overview. Pediatr. Nephrol. 2017, 32, 1123–1135. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zawadzki, J.; Grenda, R.; Madej-Pilarczyk, A.; Ciara, E. The Metabolic Consequences of Pathogenic Variant in FXYD2 Gene Encoding the Gamma Subunit of Sodium/Potassium-Transporting ATPase in Two Siblings with Sodium-Dependent Defect of Fructose, Galactose and Glucose Renal Reabsorption. Genes 2025, 16, 535. https://doi.org/10.3390/genes16050535
Zawadzki J, Grenda R, Madej-Pilarczyk A, Ciara E. The Metabolic Consequences of Pathogenic Variant in FXYD2 Gene Encoding the Gamma Subunit of Sodium/Potassium-Transporting ATPase in Two Siblings with Sodium-Dependent Defect of Fructose, Galactose and Glucose Renal Reabsorption. Genes. 2025; 16(5):535. https://doi.org/10.3390/genes16050535
Chicago/Turabian StyleZawadzki, Jan, Ryszard Grenda, Agnieszka Madej-Pilarczyk, and Elżbieta Ciara. 2025. "The Metabolic Consequences of Pathogenic Variant in FXYD2 Gene Encoding the Gamma Subunit of Sodium/Potassium-Transporting ATPase in Two Siblings with Sodium-Dependent Defect of Fructose, Galactose and Glucose Renal Reabsorption" Genes 16, no. 5: 535. https://doi.org/10.3390/genes16050535
APA StyleZawadzki, J., Grenda, R., Madej-Pilarczyk, A., & Ciara, E. (2025). The Metabolic Consequences of Pathogenic Variant in FXYD2 Gene Encoding the Gamma Subunit of Sodium/Potassium-Transporting ATPase in Two Siblings with Sodium-Dependent Defect of Fructose, Galactose and Glucose Renal Reabsorption. Genes, 16(5), 535. https://doi.org/10.3390/genes16050535