
Assembly and Comparative Analysis of the Complete Mitochondrial Genomes of Smilax glabra and Smilax zeylanica

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction of Sample and Mitogenomic Sequencing
2.2. Assembly and Annotation of the Mitogenome
2.3. Analysis of Repeat Fragments, Codon Usage Bias, and Prediction of RNA Editing Sites
2.4. Sequence Transfer Analysis
2.5. Construction of Maximum Likelihood Tree Based on the PCGs
2.6. Identification of Homologous Fragments and Collinear Analysis
3. Results
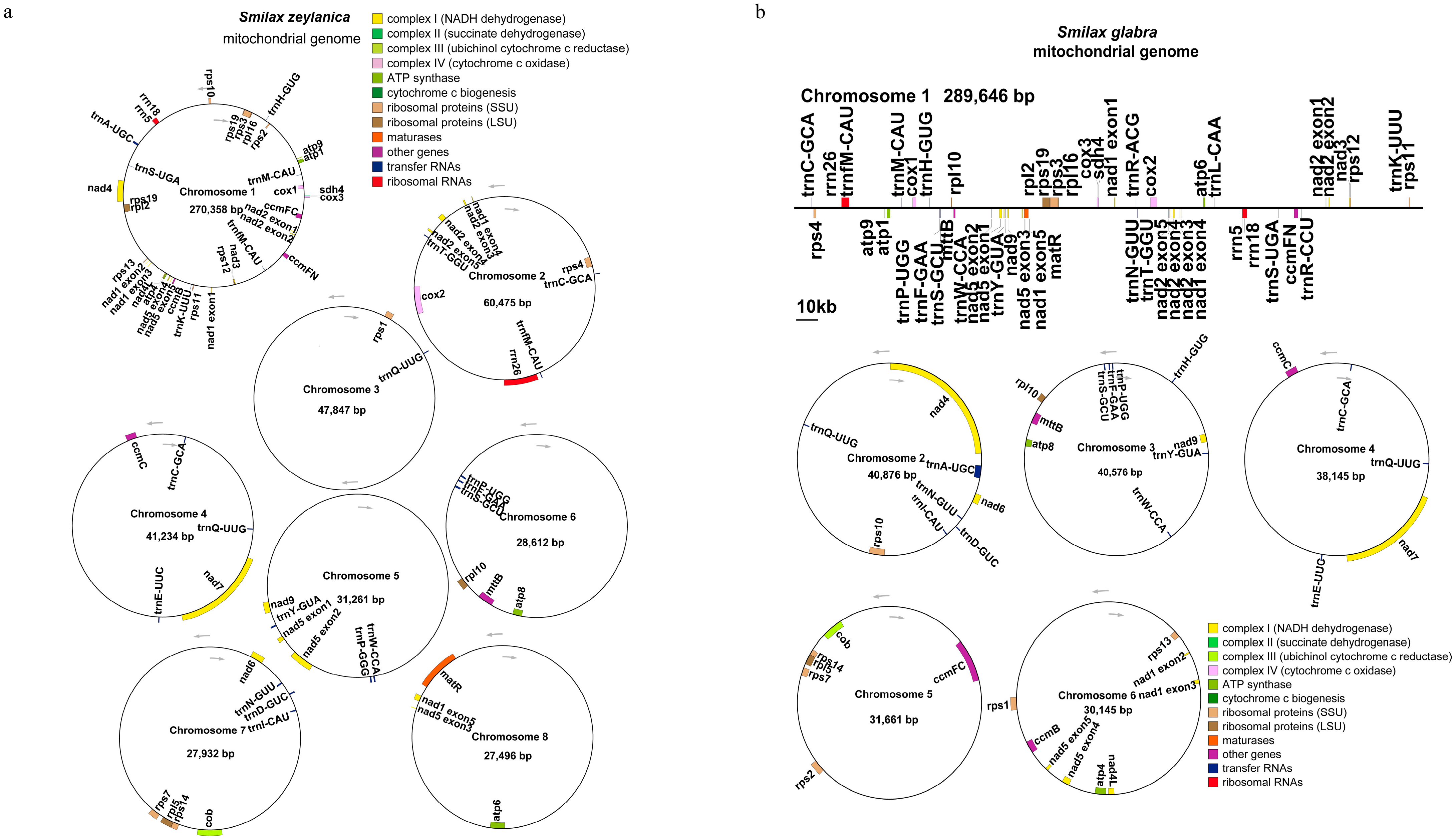
3.1. Genomic Characteristics of the Smilax Mitochondrial Genomes
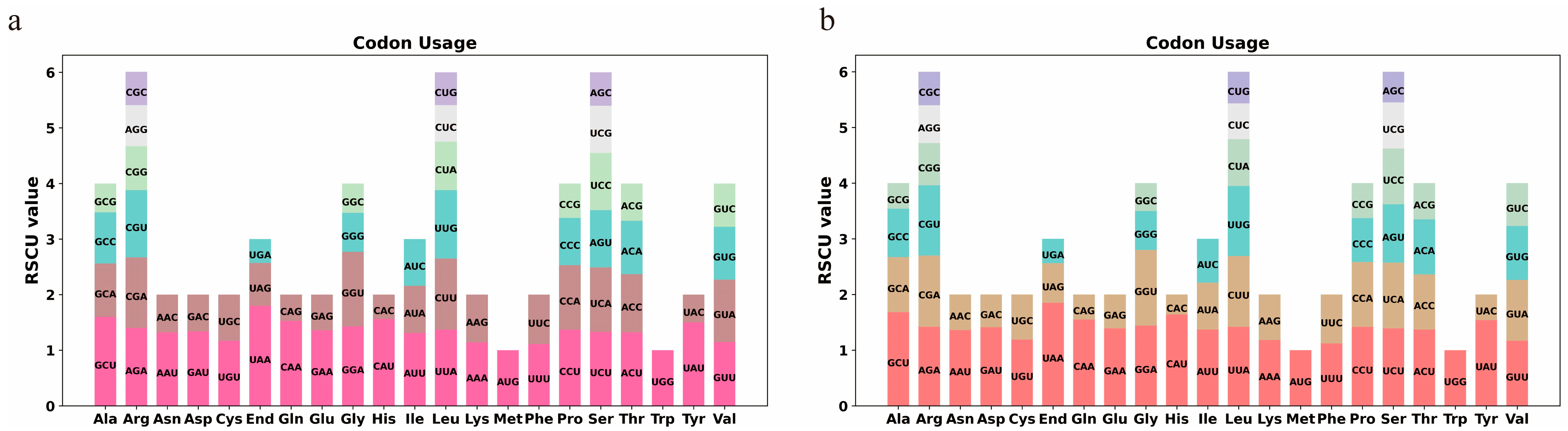
3.2. Analysis of Relative Synonymous Codon Usage
3.3. Repeat Sequence Analysis
3.4. RNA Editing Event Prediction
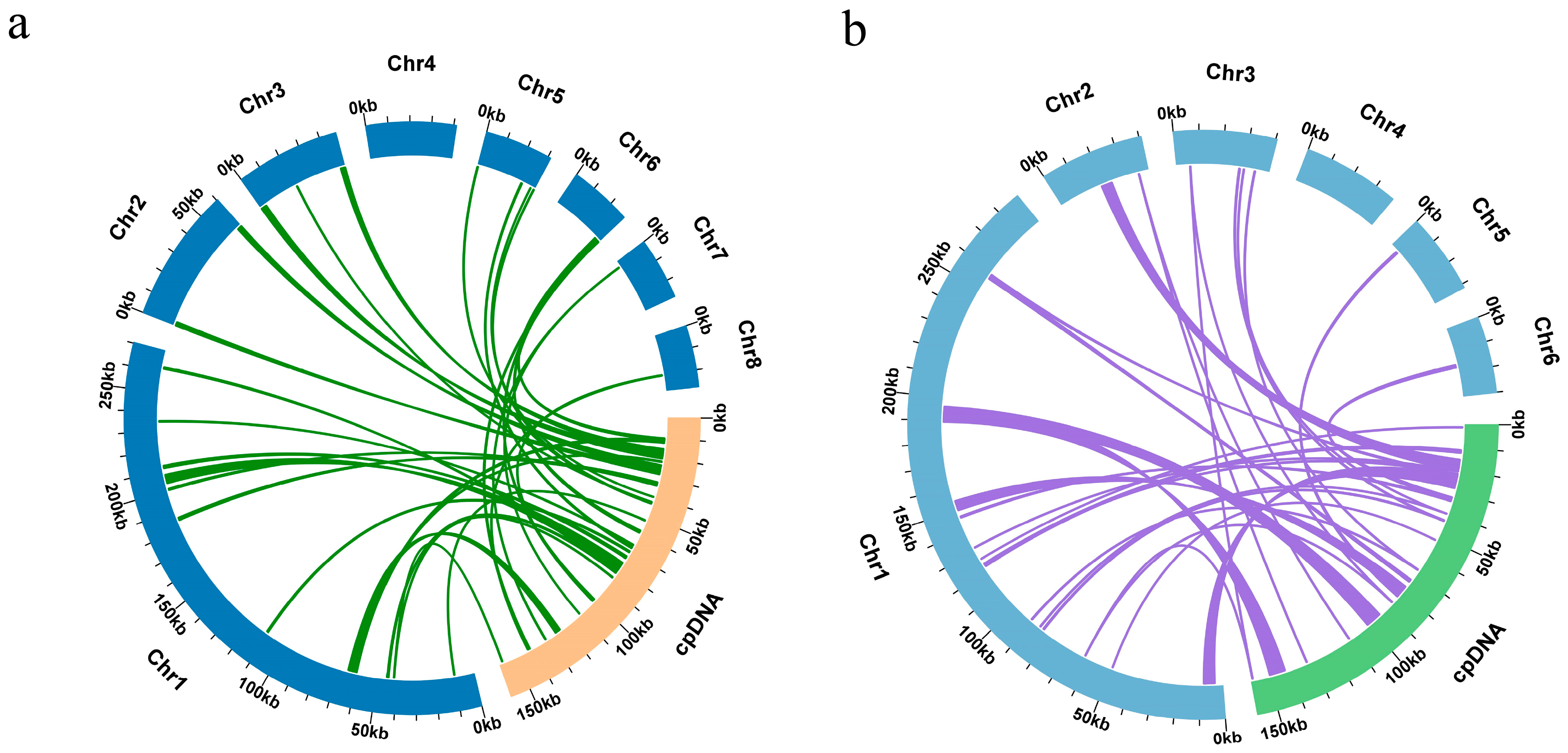
3.5. DNA Transfer from Chloroplast to Mitochondria
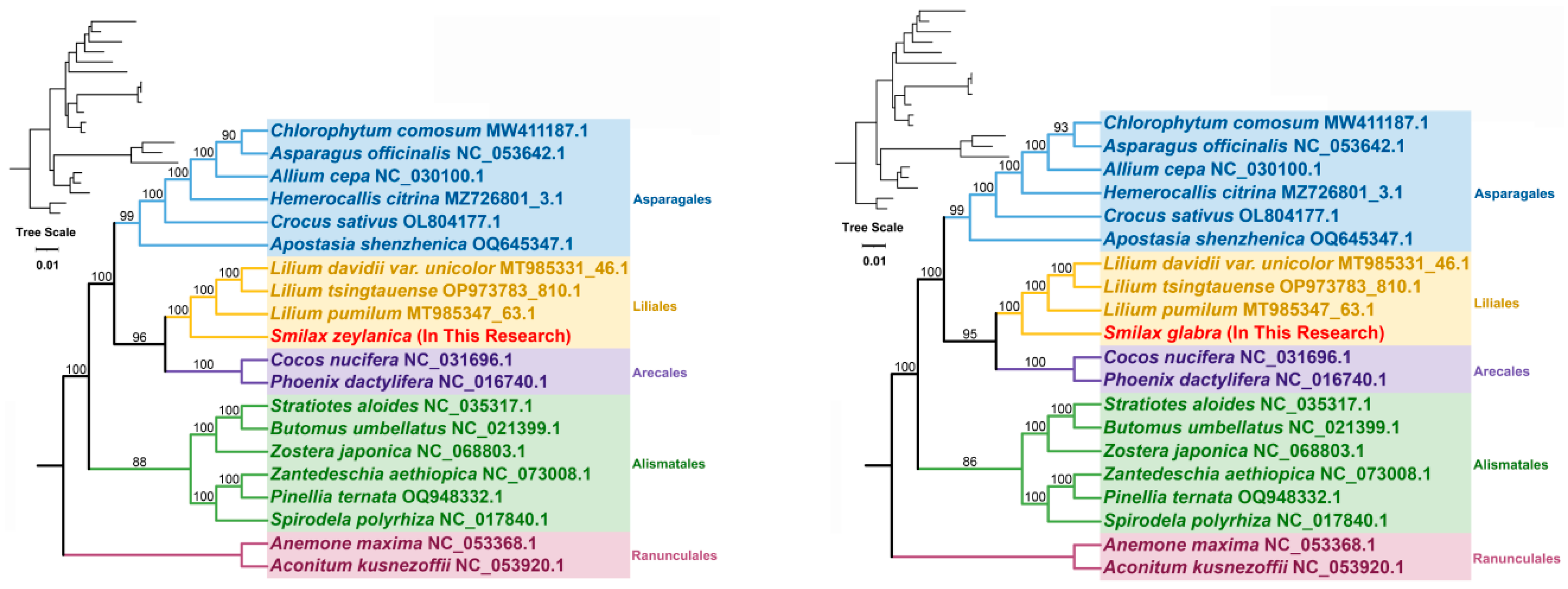
3.6. Phylogenetic Analysis Within the Mitochondrial Genome
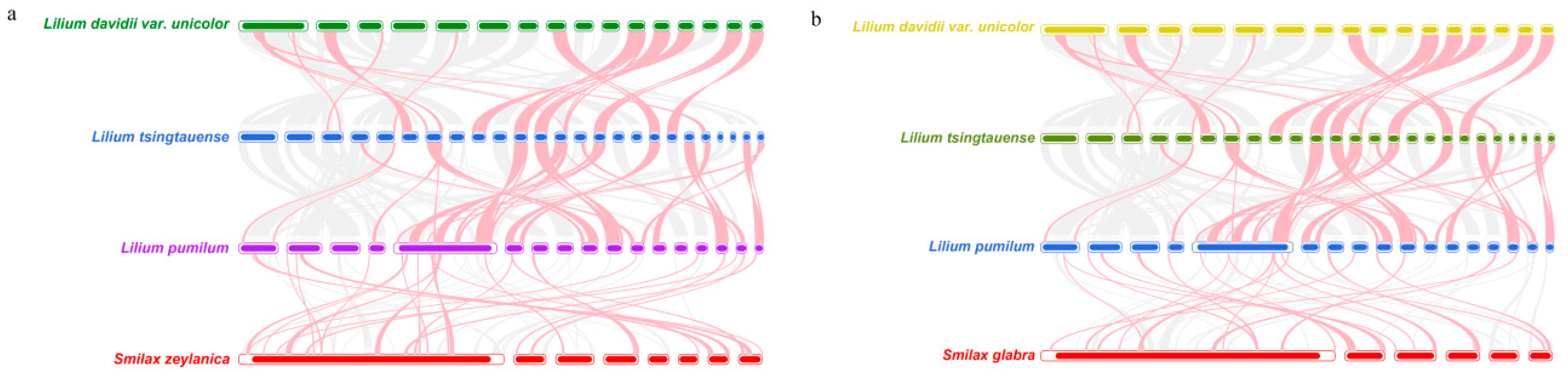
3.7. Comparative Analysis of Five Liliales Mitochondrial Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, Y.; Hu, D.; Zhong, C.; Liu, K.F.; Fang, E.; Zhang, Y.J.; Zhou, C.; Tian, L.W. Anti-inflammatory furostanol saponins from the rhizomes of Smilax china L. Steroids 2018, 140, 70–76. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Jin, W.; Zhang, W.; Zheng, G. Identification of the constituents of ethyl acetate fraction from Smilax china L. and determination of xanthine oxidase inhibitory properties. Int. J. Mol. Sci. 2023, 24, 5158. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Nisar, M.; Ebad, F.; Nadeem, S.; Saeed, M.; Khan, H.; Samiullah; Khuda, F.; Karim, N.; Ahmad, Z. Anti-inflammatory activities of Sieboldogenin from Smilax china Linn.: Experimental and computational studies. J. Ethnopharmacol. 2009, 121, 175–177. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Zhang, B.; Li, Y.L.; Ren, Z.X.; Huang, J.J.; Zhang, Z.Q.; Lin, Z.J.; Zhang, X.M. Smilax glabra Roxb.: A review of its traditional usages, phytochemical constituents, pharmacological properties, and clinical applications. Drug Des. Devel. Ther. 2022, 16, 3621–3643. [Google Scholar] [PubMed]
- Zhang, W.; Yu, L.; Yang, Q.; Zhang, J.; Wang, W.; Hu, X.; Li, J.; Zheng, G. Smilax china L. polysaccharide prevents HFD induced-NAFLD by regulating hepatic fat metabolism and gut microbiota. Phytomedicine 2024, 127, 155478. [Google Scholar] [CrossRef] [PubMed]
- Chalertpet, K.; Sangkheereeput, T.; Somjit, P.; Bankeeree, W.; Yanatatsaneejit, P. Effect of Smilax spp. and Phellinus linteus combination on cytotoxicity and cell proliferation of breast cancer cells. BMC Complement. Med. Ther. 2023, 23, 177. [Google Scholar]
- Uddin, M.N.; Ahmed, T.; Pathan, S.; Al-Amin, M.M.; Rana, M.S. Antioxidant and cytotoxic activity of stems of Smilax zeylanica in vitro. J. Basic. Clin. Physiol. Pharmacol. 2015, 26, 453–463. [Google Scholar] [CrossRef]
- Dhanya Shree, V.; Arbin, A.; Seema Noorain, G.; Sahana, B.; Prashidh Kekuda, T. Preliminary phytochemical analysis, antimicrobial and antioxidant activity of S. zeylanica L. (Smilacaceae). J. Drug Deliv. Ther. 2018, 8, 237–243. [Google Scholar] [CrossRef]
- Nath, L.; Ahmed, R.; Kashyap, B.; Sarma, H.; Bora, N.S.; Gam, S.; Deka, K.; Siangshai, S.; Sahariah, B.J.; Dutta, K.N. Exploring the anti-inflammatory and antimicrobial potential of S. zeylanica extract through in vivo and in vitro analysis. J. Pharmacogn. Phytochem. 2023, 12, 273–279. [Google Scholar] [CrossRef]
- Nazar, N.; Saxena, A.; Sebastian, A.; Slater, A.; Sundaresan, V.; Sgamma, T. Integrating DNA barcoding within an orthogonal approach for herbal product authentication: A narrative review. Phytochem. Anal. 2025, 36, 7–29. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Z.; Liu, C. Molecular identification of Gentiana macrophylla and its adulterants based on mitochondrial gene sequences. J. Ethnopharmacol. 2017, 206, 284–291. [Google Scholar]
- Liberatore, K.L.; Dukowic-Schulze, S.; Miller, M.E.; Chen, C.; Kianian, S.F. The role of mitochondria in plant development and stress tolerance. Free Radic. Biol. Med. 2016, 100, 238–256. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Xu, D.; Wang, T.; Huang, J.; Wang, Q.; Wang, Z.; Xie, Z.; Zeng, D.; Liu, X.; Fu, L. Comparative analysis of mitochondrial genomes of Stemona tuberosa Lour. reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. BMC Plant Biol. 2025, 25, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Qin, M.; Liu, X.; Qi, Y.; Ou, X.; Tang, M. De novo assembly of the mitochondrial genome of Glycyrrhiza glabra and identification of two types of homologous recombination configurations caused by repeat sequences. BMC Genom. 2025, 26, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Guo, T.; An, X. Comprehensive analysis of the multi-rings mitochondrial genome of Populus tomentosa. BMC Genom. 2025, 26, 23. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Wang, J.; Nie, L.; Tembrock, L.R.; Zou, C.; Kan, S.; Ma, X.; Wendel, J.F.; Wu, Z. Evolutionary dynamics of mitochondrial genomes and intracellular transfers among diploid and allopolyploid cotton species. BMC Biol. 2025, 23, 9. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, X.; Huang, Y.; Chen, Z.; Chen, B. Comparative mitochondrial genomics of Terniopsis yongtaiensis in Malpighiales: Structural, sequential, and phylogenetic perspectives. BMC Genom. 2024, 25, 853. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Michael, T.; Pascal, L.; Tommaso, P.; Ulbricht-Jones, E.S.; Axel, F.; Ralph, B.; Stephan, G. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, 0082. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Stefan, K.; Choudhuri, J.V.; Enno, O.; Chris, S.; Jens, S.; Robert, G. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Singh, S.; Naik, J.; Pandey, A. Genetics of Plant Organelles: Plastid and Mitochondrial Genomes. In Plant Genomics for Sustainable Agriculture; Singh, R.L., Mondal, S., Parihar, A., Singh, P.K., Eds.; Springer: Singapore, 2022; pp. 1–20. [Google Scholar]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Salamini, F.; Velasco, R.; Viola, R. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol. Biol. Evol. 2009, 26, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.K.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2013, 99, 349–364. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef]
- Miyata, T.; Kuma, K.; Iwabe, N.; Nikoh, N. Evolution of tRNA genes in the mitochondrial genome: Implications for the origin of mitochondria. J. Mol. Evol. 1998, 46, 504–512. [Google Scholar]
- Wang, D.; Wu, Y.W.; Shih, A.C.C.; Wu, C.S.; Wang, Y.N.; Chaw, S.M. Transfer of chloroplast-derived tRNA genes to the mitochondrial genome in gymnosperms and angiosperms: Evolutionary patterns and implications. Genome Biol. Evol. 2020, 12, 1789–1801. [Google Scholar]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.; Li, Y.; Qian, J.; Han, J. Chloroplast and mitochondrial DNA barcoding for identification of Gentiana macrophylla and its related species. Plant Divers. Resour. 2017, 39, 621–630. [Google Scholar]
- Madesis, P.; Ganopoulos, I.; Tsaftaris, A. Microsatellites: Evolution and Contribution. In Microsatellites; Springer: New York, NY, USA, 2013; pp. 1–20. [Google Scholar]
- Li, X.; Wang, Y.; Zhang, J.; Liu, Y.; Chen, J. Compositional bias and distribution of simple sequence repeats in plant mitochondrial genomes: Insights into AT-rich genome evolution. Front. Plant Sci. 2021, 12, 654785. [Google Scholar]
- Wynn, E.L.; Christensen, A.C. Repeats and recombination in plant mitochondrial genomes: Roles of palindromic and dispersed repeats in genome stability and evolution. Front. Plant Sci. 2019, 10, 779. [Google Scholar]
- Marechal, A.; Brisson, N.; Marechal, A. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2009, 181, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Härtel, B.; Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, C.; Yagi, Y.; Sugita, M. RNA editing and its molecular mechanism in plant organelles. Genes 2017, 8, 5. [Google Scholar] [CrossRef]
- Giegé, P.; Brennicke, A.; Binder, S. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 2005, 102, 598–603. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, G.; Liang, W.; Yu, H.; Zhang, K.; Li, L.; Feng, S.; Song, L.; Yang, C.; Wan, L.; Zeng, D.; et al. Assembly and Comparative Analysis of the Complete Mitochondrial Genomes of Smilax glabra and Smilax zeylanica. Genes 2025, 16, 450. https://doi.org/10.3390/genes16040450
Liao G, Liang W, Yu H, Zhang K, Li L, Feng S, Song L, Yang C, Wan L, Zeng D, et al. Assembly and Comparative Analysis of the Complete Mitochondrial Genomes of Smilax glabra and Smilax zeylanica. Genes. 2025; 16(4):450. https://doi.org/10.3390/genes16040450
Chicago/Turabian StyleLiao, Guojian, Wenjing Liang, Haixia Yu, Kun Zhang, Linxuan Li, Shixin Feng, Lisha Song, Cuihong Yang, Lingyun Wan, Dongqiang Zeng, and et al. 2025. "Assembly and Comparative Analysis of the Complete Mitochondrial Genomes of Smilax glabra and Smilax zeylanica" Genes 16, no. 4: 450. https://doi.org/10.3390/genes16040450
APA StyleLiao, G., Liang, W., Yu, H., Zhang, K., Li, L., Feng, S., Song, L., Yang, C., Wan, L., Zeng, D., Zhang, Z., & Wei, S. (2025). Assembly and Comparative Analysis of the Complete Mitochondrial Genomes of Smilax glabra and Smilax zeylanica. Genes, 16(4), 450. https://doi.org/10.3390/genes16040450