
Advancing Non-Invasive Prenatal Screening: A Targeted 1069-Gene Panel for Comprehensive Detection of Monogenic Disorders and Copy Number Variations

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
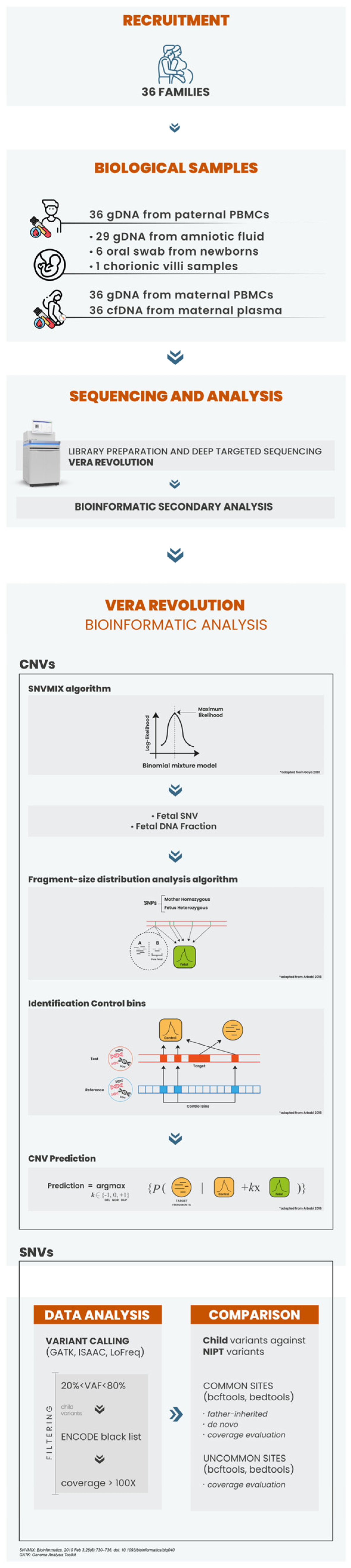
2.1. Patient Recruitment and Sample Processing
2.2. DNA Isolation and NIPS Analysis
2.3. VERA Revolution Gene Panel: Design and Selection Criteria
2.4. VERA Revolution Library Preparation
2.5. CNV Calling
2.6. Follow-Up Diagnostic Testing
3. Results
3.1. VERA Revolution Gene Panel and Depth of Analysis
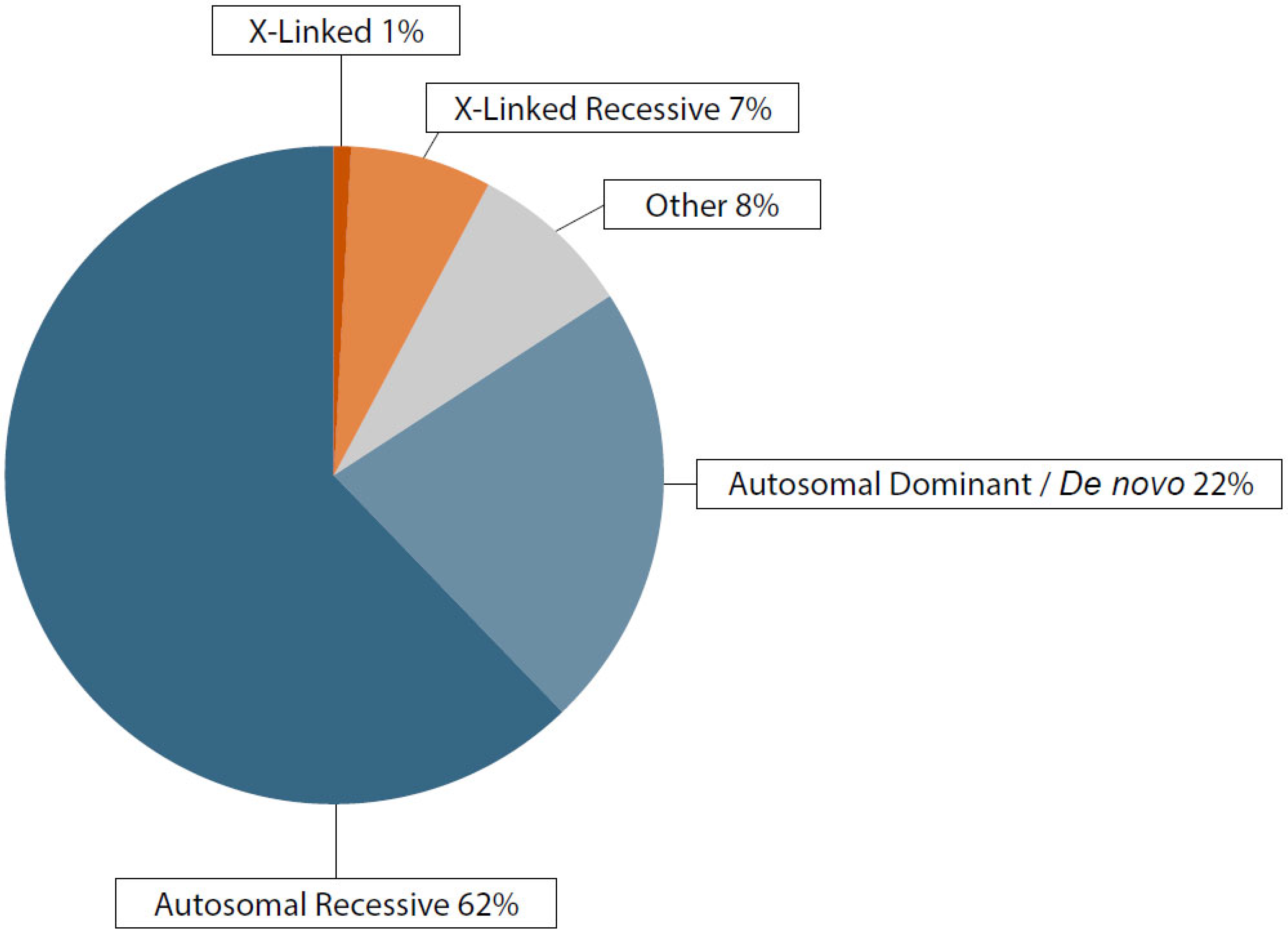
3.2. Genetic Findings
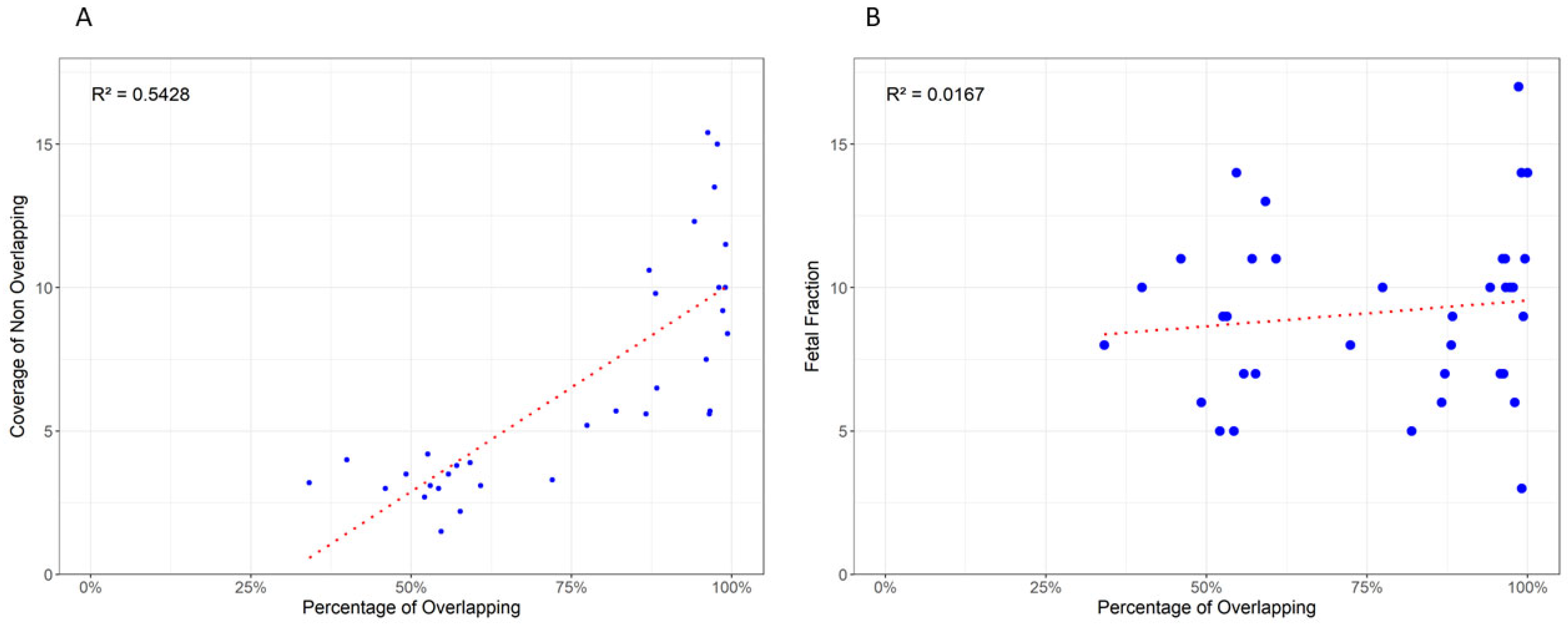
3.3. Variant Overlap and Coverage Influence
4. Discussion
4.1. Broad-Spectrum Analysis
4.2. Clinical Implications in Early Pregnancy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkins, J.C.; Padgett, C.R. Living with a Rare Disease: Psychosocial Impacts for Parents and Family Members—A Systematc Review. J. Child Fam. Stud. 2024, 33, 617–636. [Google Scholar] [CrossRef]
- Alfirevic, Z.; Navaratnam, K.; Mujezinovic, F. Amniocentesis and Chorionic Villus Sampling for Prenatal Diagnosis. Cochrane Database Syst. Rev. 2017, 2017, CD003252. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of Fetal DNA in Maternal Plasma and Serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Nectoux, J. Current, Emerging, and Future Applications of Digital PCR in Non-Invasive Prenatal Diagnosis. Mol. Diagn. Ther. 2018, 22, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Saucier, J.B.; Feng, Y.; Jiang, Y.; Sinson, J.; McCombs, A.K.; Schmitt, E.S.; Peacock, S.; Chen, S.; et al. Non-Invasive Prenatal Sequencing for Multiple Mendelian Monogenic Disorders Using Circulating Cell-Free Fetal DNA. Nat. Med. 2019, 25, 439–447. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Chan, K.C.A.; Gao, Y.; Lau, V.Y.M.; Zheng, W.; Leung, T.Y.; Foo, C.H.F.; Xie, B.; Tsui, N.B.Y.; Lun, F.M.F.; et al. Noninvasive Prenatal Diagnosis of Fetal Chromosomal Aneuploidy by Massively Parallel Genomic Sequencing of DNA in Maternal Plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef]
- Chen, C.; Li, R.; Sun, J.; Zhu, Y.; Jiang, L.; Li, J.; Fu, F.; Wan, J.; Guo, F.; An, X.; et al. Noninvasive Prenatal Testing of α-Thalassemia and β-Thalassemia through Population-Based Parental Haplotyping. Genome Med. 2021, 13, 18. [Google Scholar] [CrossRef]
- Hui, L.; Bianchi, D.W. Recent Advances in the Prenatal Interrogation of the Human Fetal Genome. Trends Genet. 2013, 29, 84–91. [Google Scholar] [CrossRef]
- Gadsbøll, K.; Petersen, O.B.; Gatinois, V.; Strange, H.; Jacobsson, B.; Wapner, R.; Vermeesch, J.R.; The NIPT-map Study Group; Vogel, I. Current Use of Noninvasive Prenatal Testing in Europe, Australia and the USA: A Graphical Presentation. Acta Obstet. Gynecol. Scand 2020, 99, 722–730. [Google Scholar] [CrossRef]
- Vora, N.L.; Langlois, S.; Chitty, L.S. Current Controversy in Prenatal Diagnosis: The Use of cfDNA to Screen for Monogenic Conditions in Low Risk Populations Is Ready for Clinical Use. Prenat. Diagn. 2024, 44, 389–397. [Google Scholar] [CrossRef]
- Rafalko, J.; Soster, E.; Caldwell, S.; Almasri, E.; Westover, T.; Weinblatt, V.; Cacheris, P. Genome-Wide Cell-Free DNA Screening: A Focus on Copy-Number Variants. Genet. Med. 2021, 23, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Dungan, J.S.; Klugman, S.; Darilek, S.; Malinowski, J.; Akkari, Y.M.N.; Monaghan, K.G.; Erwin, A.; Best, R.G.; ACMG Board of Directors. Noninvasive Prenatal Screening (NIPS) for Fetal Chromosome Abnormalities in a General-Risk Population: An Evidence-Based Clinical Guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100336. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]
- Goya, R.; Sun, M.G.F.; Morin, R.D.; Leung, G.; Ha, G.; Wiegand, K.C.; Senz, J.; Crisan, A.; Marra, M.A.; Hirst, M.; et al. SNVMix: Predicting Single Nucleotide Variants from next-Generation Sequencing of Tumors. Bioinformatics 2010, 26, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Arbabi, A.; Rampášek, L.; Brudno, M. Cell-Free DNA Fragment-Size Distribution Analysis for Non-Invasive Prenatal CNV Prediction. Bioinformatics 2016, 32, 1662–1669. [Google Scholar] [CrossRef]
- De Falco, L.; Savarese, G.; Savarese, P.; Petrillo, N.; Ianniello, M.; Ruggiero, R.; Suero, T.; Barbato, C.; Mori, A.; Ramiro, C.; et al. Clinical Experience with Genome-Wide Noninvasive Prenatal Screening in a Large Cohort of Twin Pregnancies. Genes 2023, 14, 982. [Google Scholar] [CrossRef] [PubMed]
- De Falco, L.; Vitiello, G.; Savarese, G.; Suero, T.; Ruggiero, R.; Savarese, P.; Ianniello, M.; Petrillo, N.; Bruno, M.; Legnante, A.; et al. A Case Report of a Feto-Placental Mosaicism Involving a Segmental Aneuploidy: A Challenge for Genome Wide Screening by Non-Invasive Prenatal Testing of Cell-Free DNA in Maternal Plasma. Genes 2023, 14, 668. [Google Scholar] [CrossRef]
- Scotchman, E.; Shaw, J.; Paternoster, B.; Chandler, N.; Chitty, L.S. Non-Invasive Prenatal Diagnosis and Screening for Monogenic Disorders. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 320–327. [Google Scholar] [CrossRef]
- Hanson, B.; Scotchman, E.; Chitty, L.S.; Chandler, N.J. Non-Invasive Prenatal Diagnosis (NIPD): How Analysis of Cell-Free DNA in Maternal Plasma Has Changed Prenatal Diagnosis for Monogenic Disorders. Clin. Sci. 2022, 136, 1615–1629. [Google Scholar] [CrossRef]
- Hill, M.; Twiss, P.; Verhoef, T.I.; Drury, S.; McKay, F.; Mason, S.; Jenkins, L.; Morris, S.; Chitty, L.S. Non-Invasive Prenatal Diagnosis for Cystic Fibrosis: Detection of Paternal Mutations, Exploration of Patient Preferences and Cost Analysis. Prenat. Diagn. 2015, 35, 950–958. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulou, I.; Tsoplou, P.; Mavrommatis, K.; Kroupis, C. Non-Invasive Prenatal Testing (NIPT): Limitations on the Way to Become Diagnosis. Diagnosis 2015, 2, 141–158. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| ID | Gesta-tional Week | FF | Indic-ation for NIPS | NIPS Results | Vera Revolution Panel Results | Confirmatory Test | Disease | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biological Sample | NGS Results | Karyotype | Array Results | ACMG Classification | Maternal DNA | Paternal DNA | |||||||
| 1 | 10 + 4 | 11 | MA | WT,XY | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | AF | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2), c.35del p.(Gly12ValfsTer2) | 46,XY | ND | P (PVS1, PP5, PM2) | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | Deafness, autosomal recessive 1A |
| 2 | 11 + 5 | 7 | MA | WT,XY | WT | AF | WT | 46,XY | arr(X,Y)x1,(1-22)x2 | NA | WT | WT | NA |
| 3 | 15 + 2 | 11 | AMA | dup(8)(p22q22.1), XY | WT | AF | WT | 46, XY | arr(X,Y)x1,(1-22)x2, negative for UPD | NA | WT | WT | NA |
| 4 | 14 | 10 | MA | WT,XX | g.16(222,899_223,019dup);g.16(223,093_223, 625dup) | BS | WT | 46,XX | arr[GRCh37] 16p13.3 (105,320-507,904)x3 pat 410kb; arr[GRCh37] 22q11.21-q11.22 (21,806,401-22,903,788)x1 dn 1.1 Mb | VUS/P | arr(X,1-22)x2 | arr[GRCh37] 16p13.3 (105,320-507,904)x3 | NA |
| 5 | 13 + 6 | 9 | MA | WT,XY | CFTR c.1521_1523del, p.Phe508del | AF | CFTR c.1521_1523del, p.Phe508del, c.1521_1523del, p.Phe508del | 46,XY | ND | P (PS3, PM1, PM4, PP5) | c.1521_1523del, p.Phe508del | c.1521_1523del, p.Phe508del | Cystc fibrosis |
| 6 | 12 | 8 | AMA | WT, XY | g.22(18,222,123_18,222,243del);g.22(18,222,8 5_18,222,955del) | AF | NA | 46,XY | arr[GRCh37] 22q11.21(18,877,787-21,462,353)x1 dn, 2.6 Mb | P | arr(X,1-22)x2 | WT | DiGeorge syndrome |
| 7 | 10 + 3 | 11 | Previous pregnancy with IUGR | dup(4)(p16.3p12), XX | g.4(499476_499744del);g.4(500494_500614del) | AF | WT | arr[GRCh37] 4p16.3 (48,283- 1,243,573)x1 dn, 1.2Mb | P | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | Wolf-Hirschhorn syndrome | |
| 8 | 12 + 6 | 14 | MA | border trisomy 21, XY | WT | AF | NA | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | |
| 9 | 11 + 2 | 10 | Congenital heart disease, suspicion of DiGeorge Syndrome | WT,XX | WT | AF | WT | 46,XX | arr(1-22,X)x2, negative for UPD | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 10 | 12 + 5 | 5 | MA | dup(5)(p15.31p15.2), XX | g.5(6,602,533_6,602,653dup); g.5(6,604,260_6,60 | AF | WT | 46,XX,dup(5)(p15.32p15.1) | arr[GRCh37] 5p15.32 - p15.1(5.320.850-15.346.106)x3 dn 10Mb | P | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | 5p duplication syndrome |
| 11 | 10 + 3 | 10 | MA | WT,XY | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | AF | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | NA | NA | P (PVS1, PP5, PM2) | WT | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | Deafness, autosomal recessive 1A |
| 12 | 13 + 5 | 9 | cystic igroma | WT,XY | RIT1 NM_006912.6: c.229G>T (p.Ala77Ser) | AF | RIT1 NM_006912.6: c.229G>T (p.Ala77Ser) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PM5, PP5, PM1, PP3, PM2) | WT | WT | Noonan Symdrome |
| 13 | 11 + 1 | 6 | AMA | dup(22)(q11.21q12.1), XY | WT | AF | WT | 46,XY | arr(X,Y)x1,(1-22)x2 | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA | |
| 14 | 12 | 6 | MA | WT,XX | PAH NM_000277.3: c.533A>G (p.Glu178Gly), c.1208C>T (p.Ala403Val) | AF | PAH NM_000277.3: c.533A>G (p.Glu178Gly), c.1208C>T (p.Ala403Val) | 46,XX | arr(X,1-22)x2 | P (PP5, PM1, PM5, PP3, PM2)/P (PP5, PM1, PS3, BP4) | PAH NM_000277.1: c.533A>G (p.Glu178Gly) | PAH NM_000277.1: c.1208C>T (p.Ala403Val) | Phenylchetonuria |
| 15 | 11 | 14 | Increased NT, previous pregnancy with 15q11.2 deletion | WT,XX | WT | AF | WT | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 16 | 13 + 1 | 7 | Increased NT | WT, XX | WT | AF | WT | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | |
| 17 | 12 + 5 | 17 | Increased NT | WT,XX | DHCR7 NM_001360.2 c.964-3C>G | CVS | DHCR7 NM_001360.2 c.964-3C>G | 46,XX | arr(X,1-22)x2 | VUS (PP3, PM2) | WT | DHCR7 NM_001360.2 c.964-3C>G | Smith-Lemli-Opitz Syndrome |
| 18 | 12 + 2 | 14 | Increased NT | WT,XX | WT | AF | NA | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 19 | 11 | 11 | MA | Border trisomy 18, XX | WT | AF | NA | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 20 | 12 | 5 | Increased NT | WT | WT | AF | NA | NA | NA | ||||
| 21 | 12 + 3 | 7 | MA | del(13)(q21.2q31.2), XX | WT | AF | NA | 46,XX,del(13)(q21.2q31.2) | arr[GRCh37] 13q21.2 - q31.1(60.932.642-81.657.010)x1 dn | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 22 | 12 + 4 | 7 | MA | WT,XY | GJB2 NM_004004.6: c.109G>A p.(Val37Ile); c.583A>G, p.(Met195Val) | AF | NA | 46,XY | GJB2 NM_004004.6: c.109G>A p.(Val37Ile); c.583A>G, p.(Met195Val) | P (PM1, PM5, PP3, PP5, PM2)/P (PM5, PM1, PP3, PM2, PP5) | GJB2 NM_004004.6: c.109G>A p.(Val37Ile) | GJB2 NM_004004.6: c.583A>G, p.(Met195Val) | Deafness, autosomal recessive 1A |
| 23 | 12 | 5 | PMA | trisomy 3, XX | WT | AF | NA | 46,XX | arr(X,1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 24 | 9 | 10 | WT | WT | WT | BS | NA | 46,XY | arr(X,Y)x1,(1-22)x2 | NA | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | NA |
| 25 | 14 + 5 | 10 | NA | WT | TCOF1 NM_001371623.1:c.1999dup, (p.Arg667fs) | AF | TCOF1 NM_001371623.1: c.1999dup, (p.Arg667fs) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PVS1, PM2, PP5) | arr(X,1-22)x2 | arr(X,Y)x1,(1-22)x2 | Treacher Collins |
| 26 | 10 + 3 | 8 | AMA | WT,XY | PTPN11 NM_002834.5 c.1471C>T p.(Pro491Ser) | AF | PTPN11 NM_001330437.2 c.1483C>T p.(Pro495Ser) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PP5, PM5, PM1, PP3, PM2) | WT | WT | RASopathy |
| 27 | 12 + 3 | 5 | MA | WT,XY | MCCC2 NM_022132.5 c.1015G>A, p.(Val339Met) | AF | MCCC2 NM_022132.5 c.1015G>A, p.(Val339Met) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PP5, PM1) | MCCC2 NM_022132.5 c.1015G>A, p.(Val339Met) | MCCC2 NM_022132.5 c.1015G>A, p.(Val339Met) | 3-Methylcrotonyl-CoA carboxylase 2 deficiency |
| 28 | 11 + 1 | 12 | MA | WT,XY | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | BS | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | 46,XY | arr(X,Y)x1,(1-22)x2 | p (PVS1, PP5, PM2) | WT | GJB2 NM_004004.6: c.35del p.(Gly12ValfsTer2) | Deafness, autosomal recessive 1A |
| 29 | 13 + 3 | 20 | MA | WT,XY | GBA1 NM_001005741.3: c.1226A>G (p.Asn409Ser) | BS | GBA1 NM_001005741.3: c.1226A>G (p.Asn409Ser) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PS3, PM1, BP4) | GBA1 NM_001005741.3: c.1226A>G (p.Asn409Ser) | WT | Gaucher disease type I (GD1) |
| 30 | 12 + 2 | 6 | WT,XY | PTPN11 NM_002834.5 c.5C>T, p.(Thr2Ile) | AF | PTPN11 NM_002834.5 c.5C>T, p.(Thr2Ile) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PP5, PM2, PP2, BP3) | WT | WT | RASopathy | |
| 31 | 12 + 6 | 11 | AMA | dup(2)(q31.1q32.1), XX | ABCA4 NM_000350.3 c.286A>G, p.(Asn96Asp), GBA1 NM_001005741.3 c.1226A>G, p.(Asn409Ser) | AF | ABCA4 NM_000350.3 c.286A>G, p.(Asn96Asp), GBA1 NM_001005741.3 c.1226A>G, p.(Asn409Ser) | 46,XX | arr[GRCh37] 2q31.1 - q32.1 (173.278.777-188.970.013)x3 dn, 15.7 Mb | P/PL (PM5, PP5, PM1, PP3, PM2), P/PL (PS3, PM1, BP4) | WT | ABCA4 NM_000350.3 c.286A>G, p.(Asn96Asp), GBA1 NM_001005741.3 c.1226A>G, p.(Asn409Ser) | Stargardt disease 1, Gaucher disease type I (GD1) |
| 32 | 11 + 3 | 12 | AMA | WT,XY | PAH NM_000277.3, c.143T>C p.(Leu48Ser) | BS | PAH NM_000277.3, c.143T>C p.(Leu48Ser) | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PP5, PP3, PM1, PS3, PM2) | WT | PAH NM_000277.1, c.143T>C p.(Leu48Ser) | Phenylchetonuria |
| 33 | 10 + 4 | 9 | AMA | WT,XY | PAH NM_000277.1, c.688G>A p.(Val230Ile) | BS | PAH NM_000277.1, c.688G>A p.(Val230Ile) | 46,XY | arr(X,Y)x1,(1-22)x2 | LP (PP5, PM1, PM5, PS3, PM2, BP4 | PAH NM_000277.1, c.688G>A p.(Val230Ile) | WT | Phenylchetonuria |
| 34 | 11 + 4 | 15 | AMA | WT,XY | PTPN11 NM_001330437.2:c.1504C>T, p.(Arg502Trp) | AF | PTPN11 NM_001330437.2:c.1504C>T, p.Arg502Trp | 46,XY | arr(X,Y)x1,(1-22)x2 | P (PM1, PM5, PP3, PM2) | WT | WT | RASopathy |
| 35 | 12 + 0 | 11 | MA | WT,XX | MYBPC3 NM_000256.3 c.927-9G>A | AF | MYBPC3 NM_000256.3 c.927-9G>A | 46,XX | arr(X,1-22)x2 | P (PP5, PP3, PM2) | WT | MYBPC3 NM_000256.3 c.927-9G>A | Hypertrophic cardiomyopathy |
| 36 | 12 + 0 | 18 | MA | WT,XX | GBA1 NM_001005741.3 c.1448T>C, p.(Leu483Pro) | AF | GBA1 NM_001005741.3 c.1448T>C, p.(Leu483Pro) | 46,XX | arr(X,1-22)x2 | P (PP3. PP5, PM1, PM5, PM2) | GBA1 NM_001005741.3 c.1448T>C, p.(Leu483Pro) | GBA1 NM_001005741.3 c.1448T>C, p.(Leu483Pro) | Gaucher disease type I (GD1) |
| Father-Inherited | De Novo | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Total Variants | Common Variants | Uncommon Variants | % of Common | Coverage of Uncommon | Total Variants | Common Variants | Uncommon Variants | % of Common | Coverage of Uncommon |
| 1 | 3448 | 1633 | 1815 | 47% | 3.8 | 6754 | 2446 | 4308 | 36% | 2.2 |
| 2 | 559 | 487 | 72 | 87% | 10.6 | 309 | 309 | 0 | 100% | 19.3 |
| 3 | 460 | 444 | 16 | 96% | 5.6 | 272 | 272 | 0 | 100% | 15.8 |
| 4 | 2682 | 1583 | 1099 | 59% | 4.0 | 4038 | 1613 | 2425 | 40% | 1.6 |
| 5 | 3652 | 1920 | 1732 | 52% | 4.2 | 6139 | 2018 | 4121 | 33% | 1.6 |
| 6 | 2700 | 1256 | 1444 | 46% | 3.2 | 4656 | 1587 | 3069 | 34% | 1.7 |
| 7 | 1921 | 1379 | 542 | 72% | 3.1 | 1838 | 1118 | 720 | 61% | 1.4 |
| 8 | 530 | 525 | 5 | 99% | 11.5 | 343 | 341 | 2 | 99% | 7.0 |
| 9 | 448 | 438 | 10 | 98% | 15.0 | 320 | 320 | 0 | 100% | 12.8 |
| 10 | 164 | 89 | 75 | 54% | 3.0 | 680 | 476 | 204 | 70% | 3.3 |
| 11 | 500 | 483 | 17 | 96% | 5.7 | 204 | 204 | 0 | 100% | 8.8 |
| 12 | 468 | 465 | 3 | 99% | 8.4 | 301 | 258 | 43 | 86% | 6.9 |
| 13 | 464 | 402 | 62 | 87% | 5.6 | 288 | 288 | 0 | 100% | 4.5 |
| 14 | 2720 | 1605 | 1115 | 59% | 3.5 | 4383 | 1535 | 2848 | 35% | 1.4 |
| 15 | 426 | 233 | 193 | 55% | 1.5 | 1832 | 231 | 1601 | 13% | 0.9 |
| 16 | 458 | 441 | 17 | 96% | 15.4 | 405 | 287 | 118 | 71% | 3.3 |
| 17 | 498 | 491 | 7 | 99% | 9.2 | 290 | 290 | 0 | 100% | 7.2 |
| 18 | 478 | 478 | 0 | 100% | 21.0 | 304 | 304 | 0 | 100% | 3.7 |
| 19 | 490 | 488 | 2 | 99% | 18.4 | 258 | 258 | 0 | 100% | 24.4 |
| 20 | 410 | 336 | 74 | 82% | 5.7 | 3222 | 262 | 2960 | 8% | 0.8 |
| 21 | 470 | 271 | 199 | 56% | 2.2 | 221 | 147 | 74 | 67% | 2.5 |
| 22 | 6586 | 1958 | 4628 | 30% | 3.5 | 3214 | 2112 | 1102 | 66% | 1.5 |
| 23 | 478 | 249 | 229 | 52% | 2.7 | 315 | 240 | 75 | 76% | 3.7 |
| 24 | 1952 | 1511 | 441 | 77% | 5.2 | 1952 | 1511 | 441 | 77% | 1.9 |
| 25 | 481 | 453 | 28 | 94% | 12.3 | 318 | 318 | 0 | 100% | 9.9 |
| 26 | 610 | 535 | 75 | 88% | 9.8 | 355 | 350 | 5 | 99% | 17.1 |
| 27 | 2900 | 1700 | 1200 | 59% | 3.9 | 5000 | 1800 | 3200 | 36% | 2.0 |
| 28 | 430 | 380 | 50 | 88% | 6.5 | 260 | 255 | 5 | 98% | 5.2 |
| 29 | 580 | 565 | 15 | 97% | 13.5 | 330 | 330 | 0 | 100% | 11.0 |
| 30 | 2500 | 1150 | 1350 | 46% | 3.0 | 4500 | 1500 | 3000 | 33% | 1.8 |
| 31 | 1800 | 1300 | 500 | 72% | 3.3 | 1700 | 1050 | 650 | 62% | 1.3 |
| 32 | 495 | 490 | 5 | 99% | 10.0 | 310 | 308 | 2 | 99% | 6.8 |
| 33 | 450 | 440 | 10 | 98% | 16.0 | 300 | 300 | 0 | 100% | 13.0 |
| 34 | 150 | 80 | 70 | 53% | 3.1 | 700 | 490 | 210 | 70% | 3.5 |
| 35 | 510 | 490 | 20 | 96% | 7.5 | 210 | 210 | 0 | 100% | 9.0 |
| 36 | 470 | 460 | 10 | 98% | 10.0 | 295 | 250 | 45 | 85% | 7.1 |
| Father-Inherited | De Novo | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Total Variants | Common Variants | Uncommon Variants | % of Common | Coverage of Uncommon | Total Variants | Common Variants | Uncommon Variants | % of Common | Coverage of Uncommon |
| 1 | 3448 | 1633 | 1815 | 47% | 3.8 | 6754 | 2446 | 4308 | 36% | 2.2 |
| 2 | 559 | 487 | 72 | 87% | 10.6 | 309 | 309 | 0 | 100% | 19.3 |
| 3 | 460 | 444 | 16 | 96% | 5.6 | 272 | 272 | 0 | 100% | 15.8 |
| 4 | 2682 | 1583 | 1099 | 59% | 4.0 | 4038 | 1613 | 2425 | 40% | 1.6 |
| 5 | 3652 | 1920 | 1732 | 52% | 4.2 | 6139 | 2018 | 4121 | 33% | 1.6 |
| 6 | 2700 | 1256 | 1444 | 46% | 3.2 | 4656 | 1587 | 3069 | 34% | 1.7 |
| 7 | 1921 | 1379 | 542 | 72% | 3.1 | 1838 | 1118 | 720 | 61% | 1.4 |
| 8 | 530 | 525 | 5 | 99% | 11.5 | 343 | 341 | 2 | 99% | 7.0 |
| 9 | 448 | 438 | 10 | 98% | 15.0 | 320 | 320 | 0 | 100% | 12.8 |
| 10 | 164 | 89 | 75 | 54% | 3.0 | 680 | 476 | 204 | 70% | 3.3 |
| 11 | 500 | 483 | 17 | 96% | 5.7 | 204 | 204 | 0 | 100% | 8.8 |
| 12 | 468 | 465 | 3 | 99% | 8.4 | 301 | 258 | 43 | 86% | 6.9 |
| 13 | 464 | 402 | 62 | 87% | 5.6 | 288 | 288 | 0 | 100% | 4.5 |
| 14 | 2720 | 1605 | 1115 | 59% | 3.5 | 4383 | 1535 | 2848 | 35% | 1.4 |
| 15 | 426 | 233 | 193 | 55% | 1.5 | 1832 | 231 | 1601 | 13% | 0.9 |
| 16 | 458 | 441 | 17 | 96% | 15.4 | 405 | 287 | 118 | 71% | 3.3 |
| 17 | 498 | 491 | 7 | 99% | 9.2 | 290 | 290 | 0 | 100% | 7.2 |
| 18 | 478 | 478 | 0 | 100% | 21.0 | 304 | 304 | 0 | 100% | 3.7 |
| 19 | 490 | 488 | 2 | 99% | 18.4 | 258 | 258 | 0 | 100% | 24.4 |
| 20 | 410 | 336 | 74 | 82% | 5.7 | 3222 | 262 | 2960 | 8% | 0.8 |
| 21 | 470 | 271 | 199 | 56% | 2.2 | 221 | 147 | 74 | 67% | 2.5 |
| 22 | 6586 | 1958 | 4628 | 30% | 3.5 | 3214 | 2112 | 1102 | 66% | 1.5 |
| 23 | 478 | 249 | 229 | 52% | 2.7 | 315 | 240 | 75 | 76% | 3.7 |
| 24 | 1952 | 1511 | 441 | 77% | 5.2 | 1952 | 1511 | 441 | 77% | 1.9 |
| 25 | 481 | 453 | 28 | 94% | 12.3 | 318 | 318 | 0 | 100% | 9.9 |
| 26 | 610 | 535 | 75 | 88% | 9.8 | 355 | 350 | 5 | 99% | 17.1 |
| 27 | 2900 | 1700 | 1200 | 59% | 3.9 | 5000 | 1800 | 3200 | 36% | 2.0 |
| 28 | 430 | 380 | 50 | 88% | 6.5 | 260 | 255 | 5 | 98% | 5.2 |
| 29 | 580 | 565 | 15 | 97% | 13.5 | 330 | 330 | 0 | 100% | 11.0 |
| 30 | 2500 | 1150 | 1350 | 46% | 3.0 | 4500 | 1500 | 3000 | 33% | 1.8 |
| 31 | 1800 | 1300 | 500 | 72% | 3.3 | 1700 | 1050 | 650 | 62% | 1.3 |
| 32 | 495 | 490 | 5 | 99% | 10.0 | 310 | 308 | 2 | 99% | 6.8 |
| 33 | 450 | 440 | 10 | 98% | 16.0 | 300 | 300 | 0 | 100% | 13.0 |
| 34 | 150 | 80 | 70 | 53% | 3.1 | 700 | 490 | 210 | 70% | 3.5 |
| 35 | 510 | 490 | 20 | 96% | 7.5 | 210 | 210 | 0 | 100% | 9.0 |
| 36 | 470 | 460 | 10 | 98% | 10.0 | 295 | 250 | 45 | 85% | 7.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirica, R.; Ottaiano, A.; D’Amore, L.; Ianniello, M.; Petrillo, N.; Ruggiero, R.; Castiello, R.; Mori, A.; Evangelista, E.; De Falco, L.; et al. Advancing Non-Invasive Prenatal Screening: A Targeted 1069-Gene Panel for Comprehensive Detection of Monogenic Disorders and Copy Number Variations. Genes 2025, 16, 427. https://doi.org/10.3390/genes16040427
Sirica R, Ottaiano A, D’Amore L, Ianniello M, Petrillo N, Ruggiero R, Castiello R, Mori A, Evangelista E, De Falco L, et al. Advancing Non-Invasive Prenatal Screening: A Targeted 1069-Gene Panel for Comprehensive Detection of Monogenic Disorders and Copy Number Variations. Genes. 2025; 16(4):427. https://doi.org/10.3390/genes16040427
Chicago/Turabian StyleSirica, Roberto, Alessandro Ottaiano, Luigi D’Amore, Monica Ianniello, Nadia Petrillo, Raffaella Ruggiero, Rosa Castiello, Alessio Mori, Eloisa Evangelista, Luigia De Falco, and et al. 2025. "Advancing Non-Invasive Prenatal Screening: A Targeted 1069-Gene Panel for Comprehensive Detection of Monogenic Disorders and Copy Number Variations" Genes 16, no. 4: 427. https://doi.org/10.3390/genes16040427
APA StyleSirica, R., Ottaiano, A., D’Amore, L., Ianniello, M., Petrillo, N., Ruggiero, R., Castiello, R., Mori, A., Evangelista, E., De Falco, L., Santorsola, M., Misasi, M., Savarese, G., & Fico, A. (2025). Advancing Non-Invasive Prenatal Screening: A Targeted 1069-Gene Panel for Comprehensive Detection of Monogenic Disorders and Copy Number Variations. Genes, 16(4), 427. https://doi.org/10.3390/genes16040427