

The Spectra of Pathogenic Variants and Phenotypes in a Chinese Cohort of 298 Families with Osteogenesis Imperfecta
 , , and
, , and
Abstract
1. Introduction
2. Methods
2.1. Editorial Policies and Ethical Considerations
2.2. Participants and Clinical Analysis
2.3. Nucleic Acid Isolation
2.4. Next-Generation Sequencing (NGS)
2.5. Polymerase Chain Reaction and Sanger Sequencing
2.6. Reverse Transcription-PCR and Minigene Assay
2.7. Identification of Gross Deletions and Duplications
2.8. Statistical Analysis
3. Results
3.1. Clinical Characteristics
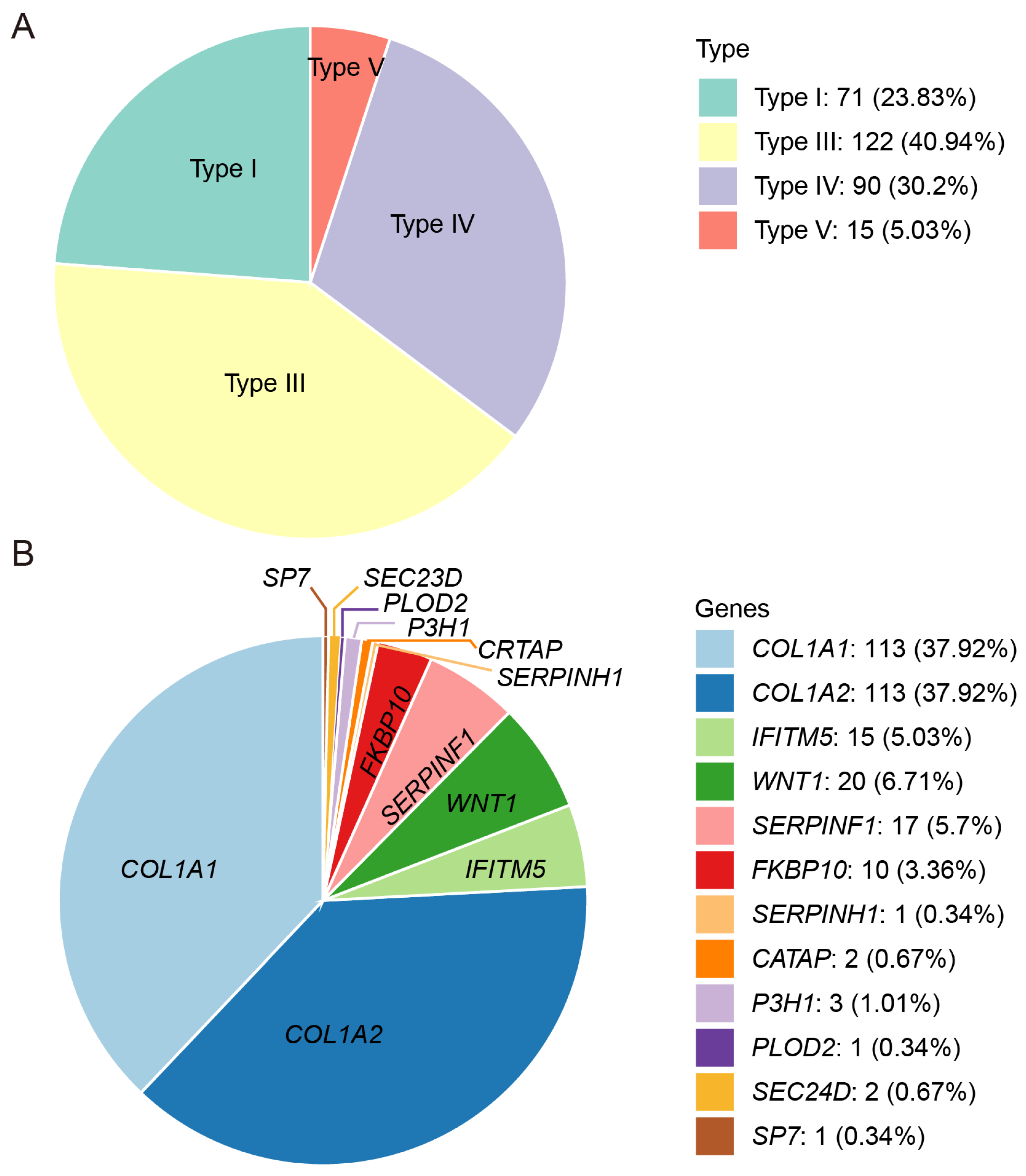
3.2. Genetic Characteristics
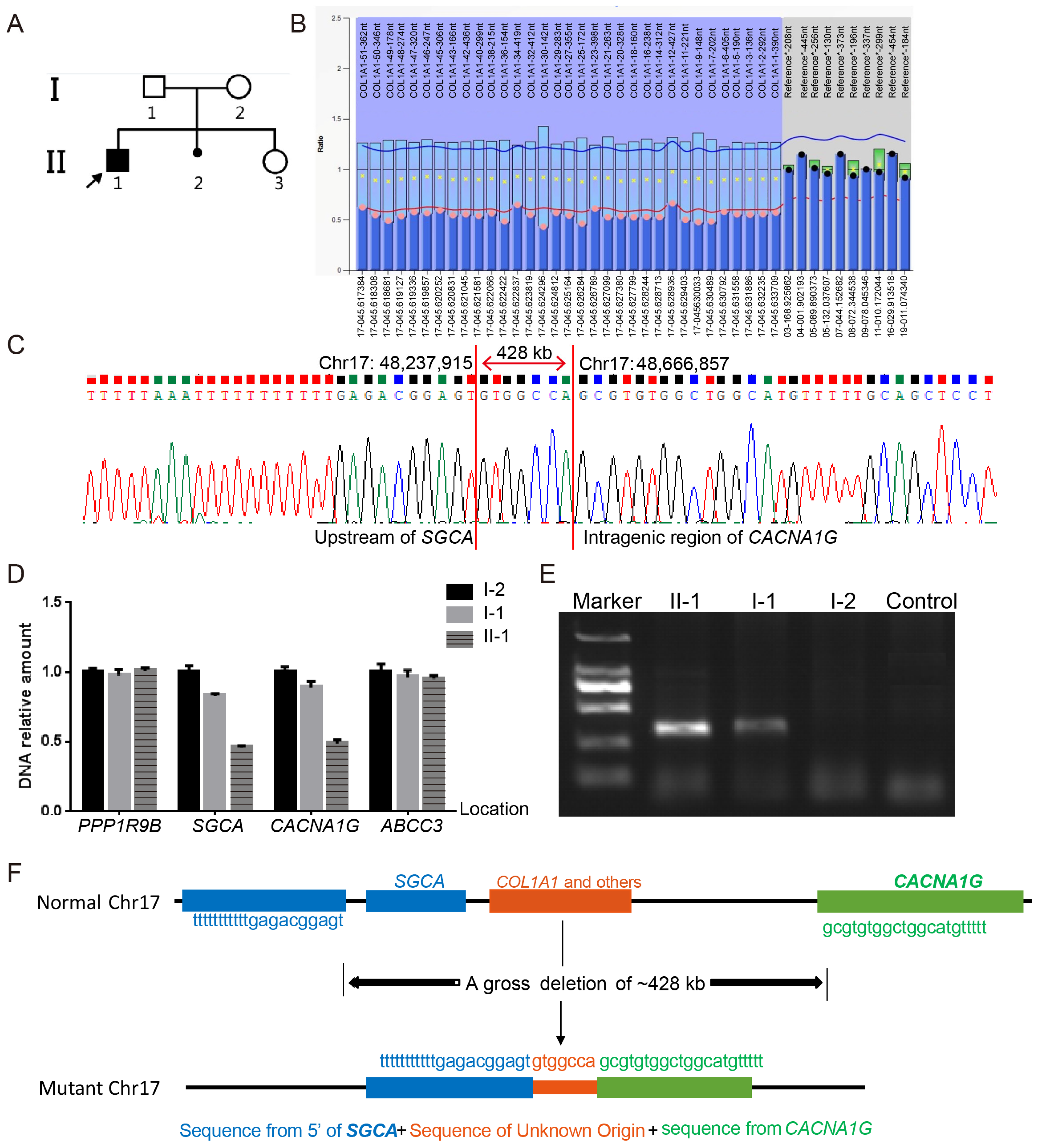
3.3. Notable Variants
3.4. Genotype–Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tournis, S.; Dede, A.D. Osteogenesis imperfecta—A clinical update. Metab. Clin. Exp. 2018, 80, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef] [PubMed]
- Fotiadou, A.N.; Calleja, M.; Hargunani, R.; Keen, R. Skeletal Manifestations of Osteogenesis Imperfecta. Semin. Musculoskelet. Radiol. 2016, 20, 279–286. [Google Scholar] [CrossRef]
- Kang, H.; Aryal, A.C.S.; Marini, J.C. Osteogenesis imperfecta: New genes reveal novel mechanisms in bone dysplasia. Transl. Res. J. Lab. Clin. Med. 2017, 181, 27–48. [Google Scholar] [CrossRef]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar]
- Marçal, F.F.; Ribeiro, E.M.; Costa, F.W.G.; Fonteles, C.S.R.; Teles, G.S.; de Barros Silva, P.G.; Chaves Junior, C.M.; Ribeiro, T.R. Dental alterations on panoramic radiographs of patients with osteogenesis imperfecta in relation to clinical diagnosis, severity, and bisphosphonate regimen aspects: A STROBE-compliant case-control study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 128, 621–630. [Google Scholar] [CrossRef]
- Carré, F.; Achard, S.; Rouillon, I.; Parodi, M.; Loundon, N. Hearing impairment and osteogenesis imperfecta: Literature review. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2019, 136, 379–383. [Google Scholar] [CrossRef]
- Rauch, F.; Glorieux, F.H. Osteogenesis imperfecta. Lancet 2004, 363, 1377–1385. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Bächinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef]
- Laine, C.M.; Joeng, K.S.; Campeau, P.M.; Kiviranta, R.; Tarkkonen, K.; Grover, M.; Lu, J.T.; Pekkinen, M.; Wessman, M.; Heino, T.J.; et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013, 368, 1809–1816. [Google Scholar] [CrossRef]
- Zhalsanova, I.Z.; Postrigan, A.E.; Valiakhmetov, N.R.; Kolesnikov, N.A.; Zhigalina, D.I.; Zarubin, A.A.; Petrova, V.V.; Minaycheva, L.I.; Seitova, G.N.; Skryabin, N.A.; et al. Case Report: A Novel Homozygous Variant of the SERPINF1 Gene in Rare Osteogenesis Imperfecta Type VI. Int. J. Mol. Sci. 2023, 24, 6672. [Google Scholar] [CrossRef] [PubMed]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef]
- Marini, J.C.; Reich, A.; Smith, S.M. Osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr. Opin. Pediatr. 2014, 26, 500–507. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, H.E.; Schwarze, U.; Pyott, S.M.; AlSwaid, A.; Al Balwi, M.; Alrasheed, S.; Pepin, M.G.; Weis, M.A.; Eyre, D.R.; Byers, P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 389–398. [Google Scholar] [CrossRef]
- Kelley, B.P.; Malfait, F.; Bonafe, L.; Baldridge, D.; Homan, E.; Symoens, S.; Willaert, A.; Elcioglu, N.; Van Maldergem, L.; Verellen-Dumoulin, C.; et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 666–672. [Google Scholar] [CrossRef]
- Leal, G.F.; Nishimura, G.; Voss, U.; Bertola, D.R.; Åström, E.; Svensson, J.; Yamamoto, G.L.; Hammarsjö, A.; Horemuzova, E.; Papadiogannakis, N.; et al. Expanding the Clinical Spectrum of Phenotypes Caused by Pathogenic Variants in PLOD2. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2018, 33, 753–760. [Google Scholar] [CrossRef]
- Xu, X.J.; Lv, F.; Song, Y.W.; Li, L.J.; Asan; Wei, X.X.; Zhao, X.L.; Jiang, Y.; Wang, O.; Xing, X.P.; et al. Novel mutations in BMP1 induce a rare type of osteogenesis imperfecta. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 489, 21–28. [Google Scholar] [CrossRef]
- Chan, E.; DeVile, C.; Ratnamma, V.S. Osteogenesis imperfecta. BJA Educ. 2023, 23, 182–188. [Google Scholar] [CrossRef]
- Jovanovic, M.; Guterman-Ram, G.; Marini, J.C. Osteogenesis Imperfecta: Mechanisms and Signaling Pathways Connecting Classical and Rare OI Types. Endocr. Rev. 2022, 43, 61–90. [Google Scholar] [CrossRef]
- Tüysüz, B.; Elkanova, L.; Uludağ Alkaya, D.; Güleç, Ç.; Toksoy, G.; Güneş, N.; Yazan, H.; Bayhan, A.I.; Yıldırım, T.; Yeşil, G.; et al. Osteogenesis imperfecta in 140 Turkish families: Molecular spectrum and, comparison of long-term clinical outcome of those with COL1A1/A2 and biallelic variants. Bone 2022, 155, 116293. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.; Moffatt, P.; Glorieux, F.H.; Rauch, F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: Diagnostic yield and mutation spectrum. Osteoporos. Int. 2016, 27, 3607–3613. [Google Scholar] [CrossRef]
- Maioli, M.; Gnoli, M.; Boarini, M.; Tremosini, M.; Zambrano, A.; Pedrini, E.; Mordenti, M.; Corsini, S.; D’Eufemia, P.; Versacci, P.; et al. Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur. J. Hum. Genet. 2019, 27, 1090–1100. [Google Scholar] [CrossRef]
- Patel, R.M.; Nagamani, S.C.; Cuthbertson, D.; Campeau, P.M.; Krischer, J.P.; Shapiro, J.R.; Steiner, R.D.; Smith, P.A.; Bober, M.B.; Byers, P.H.; et al. A cross-sectional multicenter study of osteogenesis imperfecta in North America—Results from the linked clinical research centers. Clin. Genet. 2015, 87, 133–140. [Google Scholar] [CrossRef]
- Lindahl, K.; Åström, E.; Rubin, C.J.; Grigelioniene, G.; Malmgren, B.; Ljunggren, Ö.; Kindmark, A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet 2015, 23, 1042–1050. [Google Scholar] [CrossRef]
- Li, L.; Mao, B.; Li, S.; Xiao, J.; Wang, H.; Zhang, J.; Ren, X.; Wang, Y.; Wu, Y.; Cao, Y.; et al. Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum. Mutat. 2019, 40, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cao, Y.; Zhao, F.; Mao, B.; Ren, X.; Wang, Y.; Guan, Y.; You, Y.; Li, S.; Yang, T.; et al. Validation and Classification of Atypical Splicing Variants Associated with Osteogenesis Imperfecta. Front. Genet. 2019, 10, 979. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cao, Y.; Wang, H.; Li, L.; Ren, X.; Mi, H.; Wang, Y.; Guan, Y.; Zhao, F.; Mao, B.; et al. Genotypic and Phenotypic Analysis in Chinese Cohort with Autosomal Recessive Osteogenesis Imperfecta. Front. Genet. 2020, 11, 984. [Google Scholar] [CrossRef]
- Lin, X.; Hu, J.; Zhou, B.; Zhang, Q.; Jiang, Y.; Wang, O.; Xia, W.; Xing, X.; Li, M. Genotype-phenotype relationship and comparison between eastern and western patients with osteogenesis imperfecta. J. Endocrinol. Investig. 2024, 47, 67–77. [Google Scholar] [CrossRef]
- Li, H.; Ji, C.Y.; Zong, X.N.; Zhang, Y.Q. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi = Chin. J. Pediatr. 2009, 47, 487–492. [Google Scholar]
- Sillence, D.O.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Rauch, F.; Plotkin, H.; Ward, L.; Travers, R.; Roughley, P.; Lalic, L.; Glorieux, D.F.; Fassier, F.; Bishop, N.J. Type V osteogenesis imperfecta: A new form of brittle bone disease. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2000, 15, 1650–1658. [Google Scholar] [CrossRef]
- Blouin, S.; Fratzl-Zelman, N.; Glorieux, F.H.; Roschger, P.; Klaushofer, K.; Marini, J.C.; Rauch, F. Hypermineralization and High Osteocyte Lacunar Density in Osteogenesis Imperfecta Type V Bone Indicate Exuberant Primary Bone Formation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Storoni, S.; Verdonk, S.J.E.; Zhytnik, L.; Pals, G.; Treurniet, S.; Elting, M.W.; Sakkers, R.J.B.; van den Aardweg, J.G.; Eekhoff, E.M.W.; Micha, D. From Genetics to Clinical Implications: A Study of 675 Dutch Osteogenesis Imperfecta Patients. Biomolecules 2023, 13, 281. [Google Scholar] [CrossRef] [PubMed]
- Nadyrshina, D.; Zaripova, A.; Tyurin, A.; Minniakhmetov, I.; Zakharova, E.; Khusainova, R. Osteogenesis Imperfecta: Search for Mutations in Patients from the Republic of Bashkortostan (Russia). Genes 2022, 13, 124. [Google Scholar] [CrossRef]
- Xi, L.; Zhang, H.; Zhang, Z.L. Clinical and genetic analysis in 185 Chinese probands of osteogenesis imperfecta. J. Bone Miner. Metab. 2021, 39, 416–422. [Google Scholar] [CrossRef]
- Orlando, G.; Pinedo-Villanueva, R.; Reeves, N.D.; Javaid, M.K.; Ireland, A. Physical function in UK adults with osteogenesis imperfecta: A cross-sectional analysis of the RUDY study. Osteoporos. Int. 2021, 32, 157–164. [Google Scholar] [CrossRef]
- Ohata, Y.; Takeyari, S.; Nakano, Y.; Kitaoka, T.; Nakayama, H.; Bizaoui, V.; Yamamoto, K.; Miyata, K.; Yamamoto, K.; Fujiwara, M.; et al. Comprehensive genetic analyses using targeted next-generation sequencing and genotype-phenotype correlations in 53 Japanese patients with osteogenesis imperfecta. Osteoporos. Int. 2019, 30, 2333–2342. [Google Scholar] [CrossRef]
- Binh, H.D.; Maasalu, K.; Dung, V.C.; Ngoc, C.T.; Hung, T.T.; Nam, T.V.; Nhan, L.N.; Prans, E.; Reimann, E.; Zhytnik, L.; et al. The clinical features of osteogenesis imperfecta in Vietnam. Int. Orthop. 2017, 41, 21–29. [Google Scholar] [CrossRef]
- Trancozo, M.; Moraes, M.V.D.; Silva, D.A.; Soares, J.A.M.; Barbirato, C.; Almeida, M.G.; Santos, L.R.; Rebouças, M.; Akel, A.N., Jr.; Sipolatti, V.; et al. Osteogenesis imperfecta in Brazilian patients. Genet. Mol. Biol. 2019, 42, 344–350. [Google Scholar] [CrossRef]
- Holtz, A.P.; Souza, L.T.; Ribeiro, E.M.; Acosta, A.X.; Lago, R.; Simoni, G.; Llerena, J.C., Jr.; Félix, T.M. Genetic analysis of osteogenesis imperfecta in a large Brazilian cohort. Bone 2023, 169, 116683. [Google Scholar] [CrossRef] [PubMed]
- Sałacińska, K.; Pinkier, I.; Rutkowska, L.; Chlebna-Sokół, D.; Jakubowska-Pietkiewicz, E.; Michałus, I.; Kępczyński, Ł.; Salachna, D.; Jamsheer, A.; Bukowska-Olech, E.; et al. Novel Mutations Within Collagen Alpha1(I) and Alpha2(I) Ligand-Binding Sites, Broadening the Spectrum of Osteogenesis Imperfecta—Current Insights Into Collagen Type I Lethal Regions. Front. Genet. 2021, 12, 692978. [Google Scholar] [CrossRef]
- Varenna, M.; Crotti, C.; Bonati, M.T.; Zucchi, F.; Gallazzi, M.; Caporali, R. A novel mutation in collagen gene COL1A2 associated with transient regional osteoporosis. Osteoporos. Int. 2022, 33, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, A.; Merkuryeva, E.; Zaripova, A.; Markova, T.; Nagornova, T.; Dantsev, I.; Nadyrshina, D.; Zakharova, E.; Khusainova, R. Does the c.-14C>T Mutation in the IFITM5 Gene Provide Identical Phenotypes for Osteogenesis Imperfecta Type V? Data from Russia and a Literature Review. Biomedicines 2022, 10, 2363. [Google Scholar] [CrossRef]
- Hald, J.D.; Folkestad, L.; Harsløf, T.; Lund, A.M.; Duno, M.; Jensen, J.B.; Neghabat, S.; Brixen, K.; Langdahl, B. Skeletal phenotypes in adult patients with osteogenesis imperfecta-correlations with COL1A1/COL1A2 genotype and collagen structure. Osteoporos. Int. 2016, 27, 3331–3341. [Google Scholar] [CrossRef]
- Batkovskyte, D.; Swolin-Eide, D.; Hammarsjö, A.; Sæther, K.B.; Thunström, S.; Lundin, J.; Eisfeldt, J.; Lindstrand, A.; Nordgren, A.; Åström, E.; et al. Structural Variants in COL1A1 and COL1A2 in Osteogenesis Imperfecta. Am. J. Med. Genet. Part A 2025, 197, e63935. [Google Scholar] [CrossRef]
- Ventura, L.; Verdonk, S.J.E.; Zhytnik, L.; Ridwan-Pramana, A.; Gilijamse, M.; Schreuder, W.H.; van Gelderen-Ziesemer, K.A.; Schoenmaker, T.; Micha, D.; Eekhoff, E.M.W. Dental Abnormalities in Osteogenesis Imperfecta: A Systematic Review. Calcif. Tissue Int. 2024, 115, 461–479. [Google Scholar] [CrossRef]
- Rossi, V.; Lee, B.; Marom, R. Osteogenesis imperfecta: Advancements in genetics and treatment. Curr. Opin. Pediatr. 2019, 31, 708–715. [Google Scholar] [CrossRef]
- Robinson, M.E.; Rauch, D.; Glorieux, F.H.; Rauch, F. Standardized growth charts for children with osteogenesis imperfecta. Pediatr. Res. 2023, 94, 1075–1082. [Google Scholar] [CrossRef]
- Graff, K.; Syczewska, M. Developmental charts for children with osteogenesis imperfecta, type I (body height, body weight and BMI). Eur. J. Pediatr. 2017, 176, 311–316. [Google Scholar] [CrossRef]
- Barber, L.A.; Abbott, C.; Nakhate, V.; Do, A.N.D.; Blissett, A.R.; Marini, J.C. Longitudinal growth curves for children with classical osteogenesis imperfecta (types III and IV) caused by structural pathogenic variants in type I collagen. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1233–1239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individuals (n) | Age (Year) Mean ± SD | Height Z-Score Mean ± SD | Age at First Fracture (Year) Mean ± SD | Number of Fractures (n) Mean ± SD | Frequency of Fractures (n/Year) Mean ± SD | Scoliosis (Yes) % | Walking Ability (Yes)% | Blue Sclerae (Yes) % | DI (Yes) % | Hearing Loss (Yes) % | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 298 | 15.385 ± 11.703 | −4.454 ± 4.085 | 5.54 ± 7.494 | 17.88 ± 23.756 | 2.603 ± 3.286 | 24.643 | 48.059 | 82.143 | 66.182 | 16.116 |
| Female | 151 | 15.9 ± 11.441 | −5.067 ± 4.39 | 5.468 ± 7.658 | 18.928 ± 25.447 | 2.212 ± 2.541 | 27.586 | 48.039 | 83.453 | 67.153 | 18.033 |
| Male | 147 | 14.859 ± 11.982 | −3.802 ± 3.638 | 5.58 ± 7.337 | 16.833 ± 21.977 | 2.99 ± 3.858 | 21.678 | 47.573 | 80.851 | 65.217 | 14.167 |
| p value | 0.444 | 0.012 | 0.899 | 0.46 | 0.046 | 0.245 | 0.947 | 0.57 | 0.734 | 0.413 | |
| Children | 195 | 8.053 ± 4.302 | −3.237 ± 3.003 | 2.457 ± 2.896 | 13.284 ± 19.027 | 3.101 ± 3.811 | 19.681 | 54.412 | 78.261 | 61.364 | 7.895 |
| Adult | 102 | 29.402 ± 7.978 | −6.629 ± 4.814 | 11.345 ± 9.759 | 26.647 ± 28.976 | 1.646 ± 1.528 | 34 | 34.783 | 89.583 | 74.747 | 30 |
| p value | - | <0.001 | <0.001 | <0.001 | <0.001 | 0.007 | 0.008 | 0.019 | 0.024 | <0.001 | |
| Type I | 71 | 11.754 ± 12.137 | −2.115 ± 2.8 | 5.455 ± 9.337 | 4.682 ± 2.763 | 1.888 ± 2.387 | 5.97 | 90.244 | 93.939 | 54.545 | 10.909 |
| Type III | 122 | 19.721 ± 10.907 | −6.828 ± 4.456 | 5.793 ± 6.679 | 30.935 ± 31.206 | 2.886 ± 3.52 | 40.336 | 24.211 | 83.051 | 81.034 | 21.905 |
| Type IV | 90 | 12.778 ± 10.554 | −2.958 ± 2.125 | 5.123 ± 6.385 | 10.783 ± 8.19 | 2.794 ± 3.634 | 16.092 | 54.386 | 72.84 | 58.974 | 11.765 |
| Type V | 15 | 12.7 ± 12.349 | −2.627 ± 3.252 | 5.997 ± 10.664 | 11.214 ± 9.2 | 2.348 ± 2.19 | 33.333 | 58.333 | 73.333 | 40 | 14.286 |
| p value | 0.426 | 0.155 | 0.938 | 0.042 | 0.121 | <0.001 | <0.001 | 0.008 | <0.001 | 0.196 | |
| Type I | |||||||||||
| I A | 36 | 14.224 ± 13.185 | −2.736 ± 3.297 | 7.802 ± 11.717 | 5.111 ± 3.059 | 1.754 ± 2.127 | 2.857 | 100 | 97.143 | 0 | 13.793 |
| I B | 30 | 9.556 ± 11.087 | −1.369 ± 1.881 | 2.577 ± 3.96 | 4.214 ± 2.378 | 2.011 ± 2.752 | 10 | 80 | 89.655 | 100 | 7.692 |
| p value | 0.123 | 0.056 | 0.016 | 0.192 | 0.686 | 0.232 | 0.035 | 0.218 | <0.001 | 0.469 | |
| Gene | Nucleic Acid Change | Distribution of Variants | Amino Acid Change | Effects |
|---|---|---|---|---|
| COL1A1 | c.122dupT | exon 2 | Glu42Thrfs*8 | Frameshift |
| c.182G>A | exon 2 | Cys61Tyr | Missense | |
| c.261dupC | exon 2 | Glu88Argfs*81 | Frameshift | |
| c.370-1G>C | intron 4 | - | Splicing | |
| c.545delG | exon 7 | Gly182Valfs*83 | Frameshift | |
| c.868G>C | exon 13 | Gly290Arg | Splicing | |
| c.904-2A>T | intron 13 | - | Splicing | |
| c.1201G>C | exon 19 | Gly401Arg | Missense | |
| c.1299+5G>T | intron 19 | - | Splicing | |
| c.1886G>C | exon 28 | Gly629Ala | Missense | |
| c.2354delG | exon 34 | Gly785Valfs*323 | Frameshift | |
| c.2714G>C | exon 39 | Gly905Ala | Missense | |
| c.2795delT | exon 39 | Gly926Valfs*182 | Frameshift | |
| c.2929G>A | exon 40 | Gly977Ser | Missense | |
| c.2938-1G>C | intron 40 | - | Splicing | |
| c.3068C>T | exon 42 | Ser1023Phe | Missense | |
| c.3008delC | exon 42 | Pro1003Leufs*105 | Frameshift | |
| c.3433G>A | exon 47 | Gly1145Ser | Missense | |
| c.3679dupC | exon 48 | Arg1227Profs*2 | Frameshift | |
| c.4309_4357dup | exon 51 | Glu1453Alafs*114 | Frameshift | |
| c.4325_4326del | exon 51 | Val1442Glyfs*108 | Frameshift | |
| chr17: 48237915_48666858 | whole gene | - | Gross deletion | |
| chr17: 48286224_48265650 | exon 1-43 | - | Gross duplication | |
| COL1A2 | c.739-1g>c | intron 15 | - | Splicing |
| c.792G>T | exon 16 | Lys264Asn | Splicing | |
| c.812G>T | exon 17 | Gly271Val | Missense | |
| c.866G>A | exon 17 | Gly289Asp | Missense | |
| c.962_979dupCCCGGCCTCCCTGGACCC | exon 19 | Arg327Profs*1047 | Frameshift | |
| c.2133+8a>c | intron 35 | - | Splicing | |
| c.2350-87_2350-124del | intron 38 | - | Splicing | |
| c.1207G>A | exon 22 | Gly403Ser | Missense | |
| c.1297_1300del5 | exon 23 | 433_435del | In-frame | |
| c.1324G>C | exon 23 | Gly442Arg | Missense | |
| c.1657G>C | exon 28 | Gly553Arg | Missense | |
| c.2179G>C | exon 36 | Gly727Arg | Missense | |
| c.2359G>A | exon 39 | Gly787Ser | Missense | |
| c.2405G>C | exon 40 | Gly802Ala | Missense | |
| c.2415_2423dupCCCTCCTGG | exon 40 | Pro810_Pro812dup | In-frame | |
| c.2783G>T | exon 43 | Gly928Val | Missense | |
| c.2846G>C | exon 44 | Gly949Ala | Missense | |
| WNT1 | c.403G>T | exon 3 | Val135Phe | Missense |
| c.918-3C>G | intron 4 | - | Splicing | |
| SERPINF1 | c.151G>A | exon 2 | Val51Met | Splicing |
| FKBP10 | c.812_813del2 | exon 5 | Glu271Aspfs*101 | Missense |
| g.41818629_41824066del5438 | from intron 4 | - | Splicing | |
| CRTAP | c.471+3delA | intron 1 | - | Splicing |
| SP7 | c.38G>T | exon 2 | Gly13Val | Missense |
| Individuals (n) | Age (Year) Mean ± SD | Height Z-Score Mean ± SD | Age at First Fracture (Year) Mean ± SD | Number of Fractures (n) Mean ± SD | Frequency of Fractures (n/Year) Mean ± SD | Scoliosis (Yes) % | Walking Ability (Yes) % | Blue Sclerae (Yes) % | DI (yes) % | Hearing Loss (Yes) % | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| COL1A1 | 113 | 16.875 ± 12.161 | −4.119 ± 4.516 | 5.818 ± 7.131 | 15.84 ± 20.324 | 2.354 ± 3.312 | 21.905 | 55.556 | 96.078 | 63.366 | 24.444 |
| COL1A2 | 113 | 17.425 ± 11.61 | −5.054 ± 3.81 | 6.53 ± 8.404 | 19.16 ± 24.892 | 2.052 ± 1.787 | 24.107 | 45.679 | 89.815 | 77.778 | 14.433 |
| IFITM5 | 15 | 12.7 ± 12.349 | −2.627 ± 3.252 | 5.997 ± 10.664 | 11.214 ± 9.2 | 2.348 ± 2.19 | 33.333 | 58.333 | 73.333 | 40 | 14.286 |
| WNT1 | 20 | 6.708 ± 6.617 | −5.065 ± 3.653 | 1.837 ± 2.286 | 12.889 ± 14.05 | 3.391 ± 3.563 | 25 | 30.769 | 40 | 30 | 5.882 |
| SERPINF1 | 17 | 7.882 ± 5.372 | −3.386 ± 2.513 | 2.186 ± 2.874 | 32.794 ± 38.085 | 7.038 ± 6.988 | 18.75 | 27.273 | 50 | 75 | 0 |
| FKBP10 | 10 | 12.95 ± 9.051 | −5.965 ± 4.414 | 6.183 ± 5.636 | 18.722 ± 31.214 | 1.792 ± 1.355 | 40 | 25 | 11.111 | 71.429 | 0 |
| p value | 0.147 | 0.08 | 0.078 | <0.001 | 0.759 | 0.19 | <0.001 | <0.001 | 0.086 | ||
| AD OI | 241 | 16.873 ± 11.917 | −4.476 ± 4.156 | 6.171 ± 7.971 | 17.129 ± 22.201 | 2.208 ± 2.609 | 23.707 | 50.909 | 91.556 | 68.75 | 18.905 |
| AR OI | 57 | 9.118 ± 8.272 | −4.359 ± 3.798 | 2.705 ± 3.741 | 21.056 ± 29.47 | 4.238 ± 4.939 | 28.571 | 35 | 43.636 | 54.902 | 2.439 |
| p value | <0.001 | 0.858 | <0.001 | 0.361 | 0.004 | 0.448 | 0.071 | <0.001 | 0.059 | 0.009 | |
| collagen I quantitative variant | 32 | 16.094 ± 12.721 | −1.128 ± 2.011 | 5.978 ± 6.402 | 11.823 ± 12.518 | 1.794 ± 1.468 | 16.129 | 78.947 | 100 | 50 | 22.222 |
| collagen I structural variant | 162 | 17.95 ± 11.83 | −5.52 ± 4.324 | 6.449 ± 8.173 | 19.44 ± 25.315 | 2.233 ± 2.87 | 26.452 | 43.243 | 90.132 | 76 | 18.657 |
| p value | 0.45 | <0.001 | 0.724 | 0.014 | 0.218 | 0.224 | 0.004 | 0.068 | 0.004 | 0.668 | |
| collagen I Gly substitution | 160 | 18.39 ± 12.393 | −5.376 ± 4.361 | 6.787 ± 8.441 | 18.455 ± 24.183 | 2.113 ± 2.581 | 27.097 | 46.364 | 91.447 | 73.826 | 17.91 |
| collagen I other substitution | 66 | 14.167 ± 9.945 | −2.486 ± 2.751 | 4.67 ± 5.675 | 15.105 ± 18.549 | 2.415 ± 2.788 | 12.903 | 60.465 | 96.552 | 63.333 | 22.642 |
| p value | 0.008 | <0.001 | 0.034 | 0.28 | 0.467 | 0.025 | 0.117 | 0.199 | 0.131 | 0.46 | |
| α1(I) chain structural variant | 64 | 19.095 ± 12.124 | −5.928 ± 4.975 | 6.631 ± 8.139 | 19.754 ± 24.708 | 2.605 ± 4.131 | 29.31 | 40.476 | 93.103 | 77.193 | 26.923 |
| α2(I) chain structural variant | 98 | 17.214 ± 11.641 | −5.262 ± 3.863 | 6.337 ± 8.235 | 19.245 ± 25.813 | 2.002 ± 1.654 | 24.742 | 44.928 | 88.298 | 75.269 | 13.415 |
| p value | 0.331 | 0.392 | 0.828 | 0.903 | 0.289 | 0.533 | 0.646 | 0.335 | 0.789 | 0.05 | |
| α1(I) chain Gly substitution | 68 | 19.493 ± 12.961 | −5.471 ± 4.971 | 6.969 ± 8.12 | 18.648 ± 23.72 | 2.384 ± 3.55 | 29.688 | 46.667 | 95.312 | 70.968 | 23.214 |
| α2(I) chain Gly substitution | 92 | 17.587 ± 11.971 | −5.306 ± 3.88 | 6.658 ± 8.701 | 18.317 ± 24.639 | 1.923 ± 1.583 | 25.275 | 46.154 | 88.636 | 75.862 | 14.103 |
| p value | 0.346 | 0.828 | 0.82 | 0.933 | 0.337 | 0.543 | 0.958 | 0.146 | 0.503 | 0.175 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Ren, X.; Cao, Y.; Mi, H.; Han, M.; Li, L.; Jiang, C.; Ye, Y.; Zheng, C.; Zhao, B.; et al. The Spectra of Pathogenic Variants and Phenotypes in a Chinese Cohort of 298 Families with Osteogenesis Imperfecta. Genes 2025, 16, 416. https://doi.org/10.3390/genes16040416
Zhou S, Ren X, Cao Y, Mi H, Han M, Li L, Jiang C, Ye Y, Zheng C, Zhao B, et al. The Spectra of Pathogenic Variants and Phenotypes in a Chinese Cohort of 298 Families with Osteogenesis Imperfecta. Genes. 2025; 16(4):416. https://doi.org/10.3390/genes16040416
Chicago/Turabian StyleZhou, Siji, Xiuzhi Ren, Yixuan Cao, Huan Mi, Mingchen Han, Lulu Li, Chendan Jiang, Yuqian Ye, Chaoqun Zheng, Binshan Zhao, and et al. 2025. "The Spectra of Pathogenic Variants and Phenotypes in a Chinese Cohort of 298 Families with Osteogenesis Imperfecta" Genes 16, no. 4: 416. https://doi.org/10.3390/genes16040416
APA StyleZhou, S., Ren, X., Cao, Y., Mi, H., Han, M., Li, L., Jiang, C., Ye, Y., Zheng, C., Zhao, B., Yang, T., Wu, N., Li, Z., Wu, L., & Zhao, X. (2025). The Spectra of Pathogenic Variants and Phenotypes in a Chinese Cohort of 298 Families with Osteogenesis Imperfecta. Genes, 16(4), 416. https://doi.org/10.3390/genes16040416