
GRM1 as a Candidate Gene for Buffalo Fertility: Insights from Genome-Wide Association Studies and Its Role in the FOXO Signaling Pathway

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Phenotypes and Animal Resources
2.3. Sample Collection and Sequencing
2.4. Alignments and Variant Identification
2.5. Variation Filtering
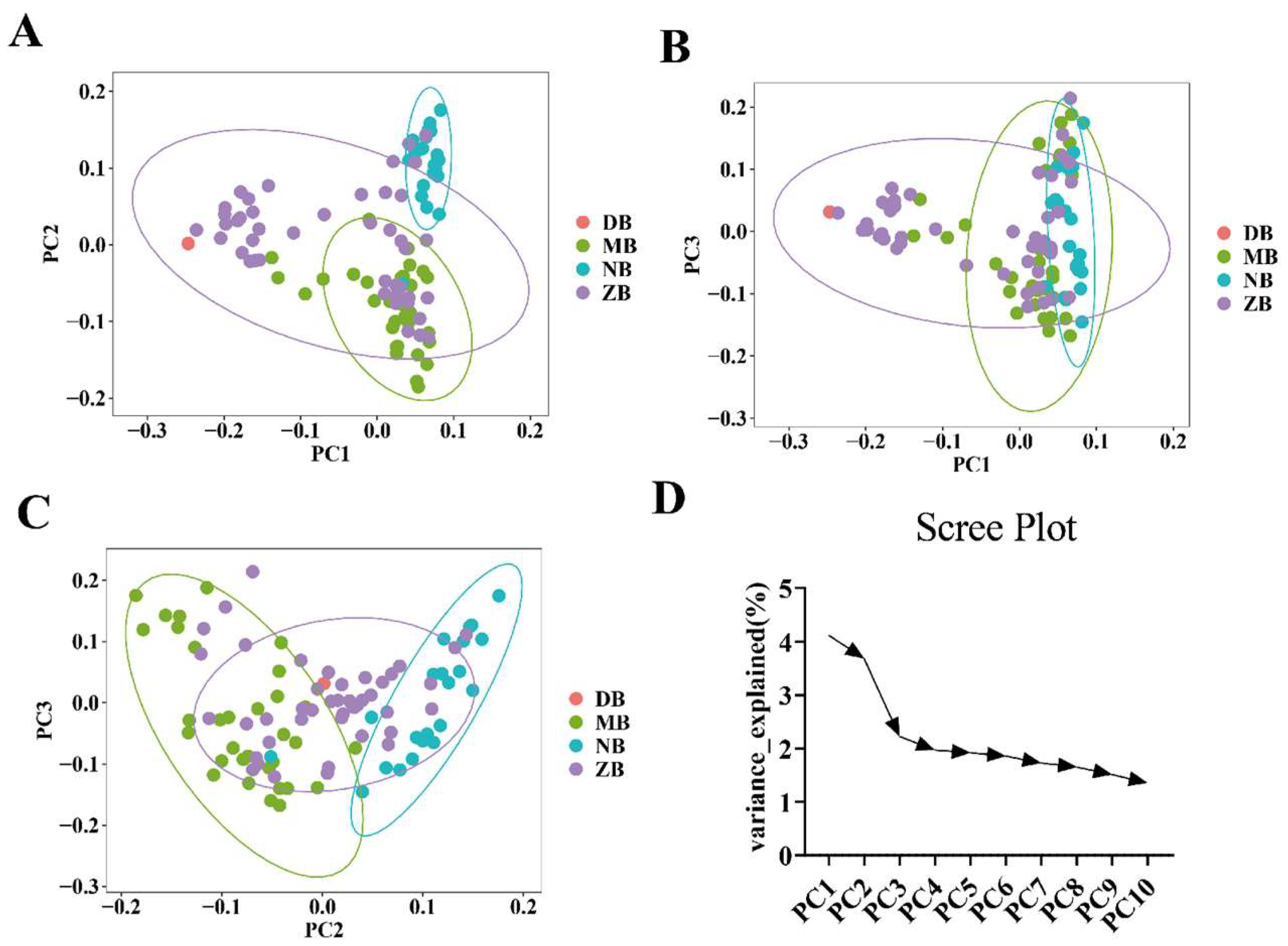
2.6. Principal Component Analysis
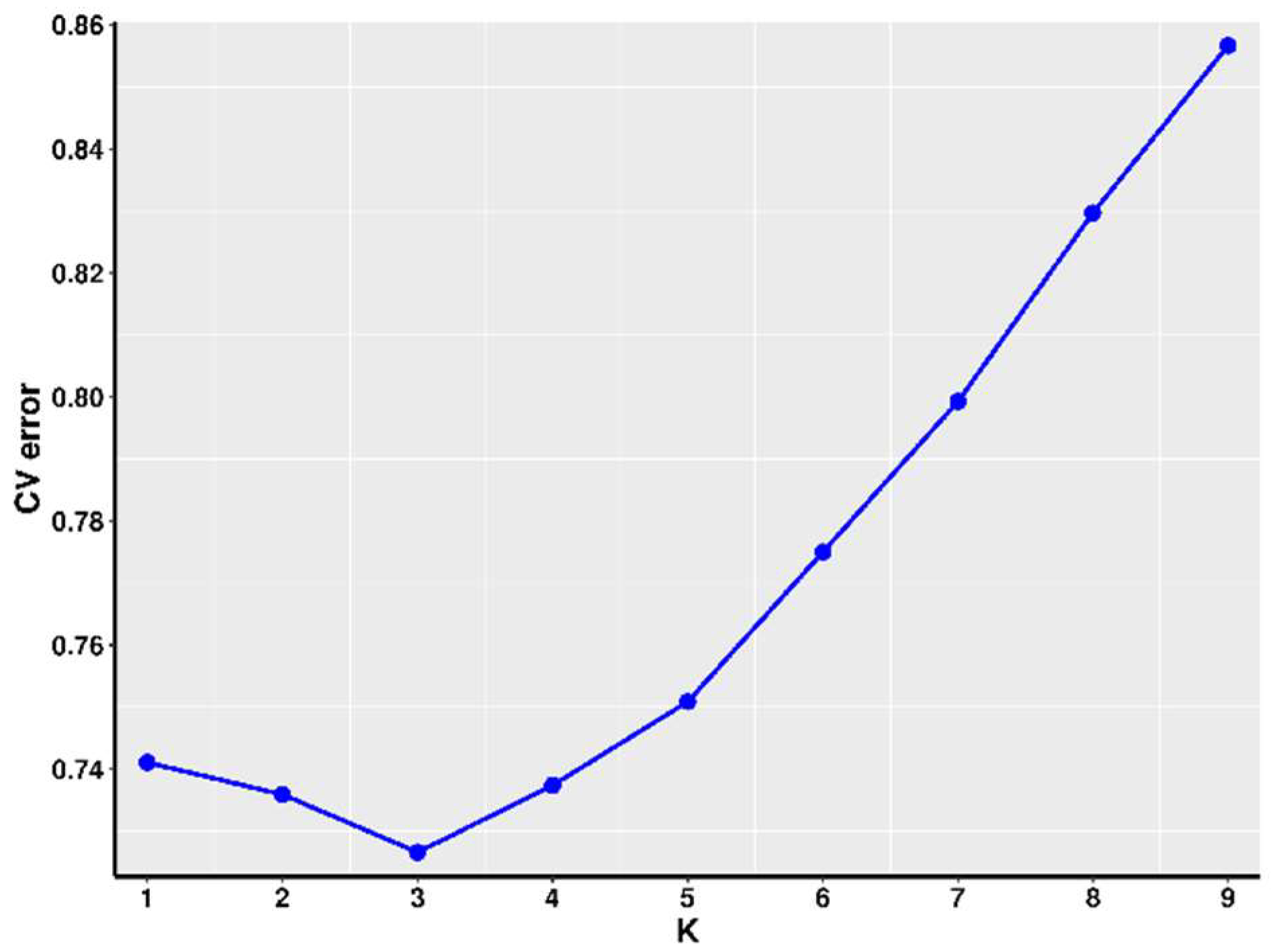
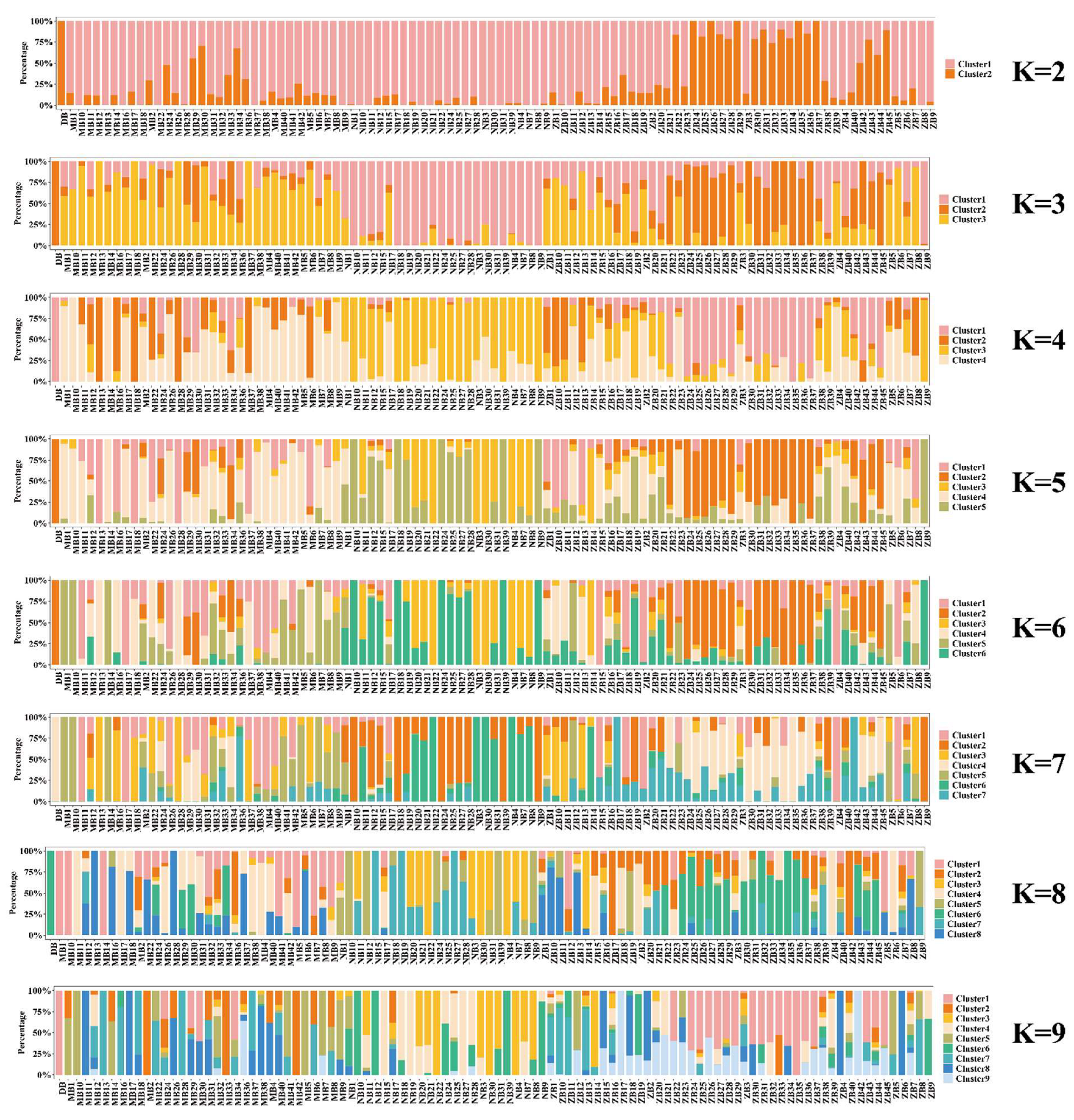
2.7. Population Structure Analysis
2.8. Genome-Wide Association Mapping
2.9. Pathway Enrichment and Protein–Protein Interaction
2.10. Statistical Analysis
3. Results
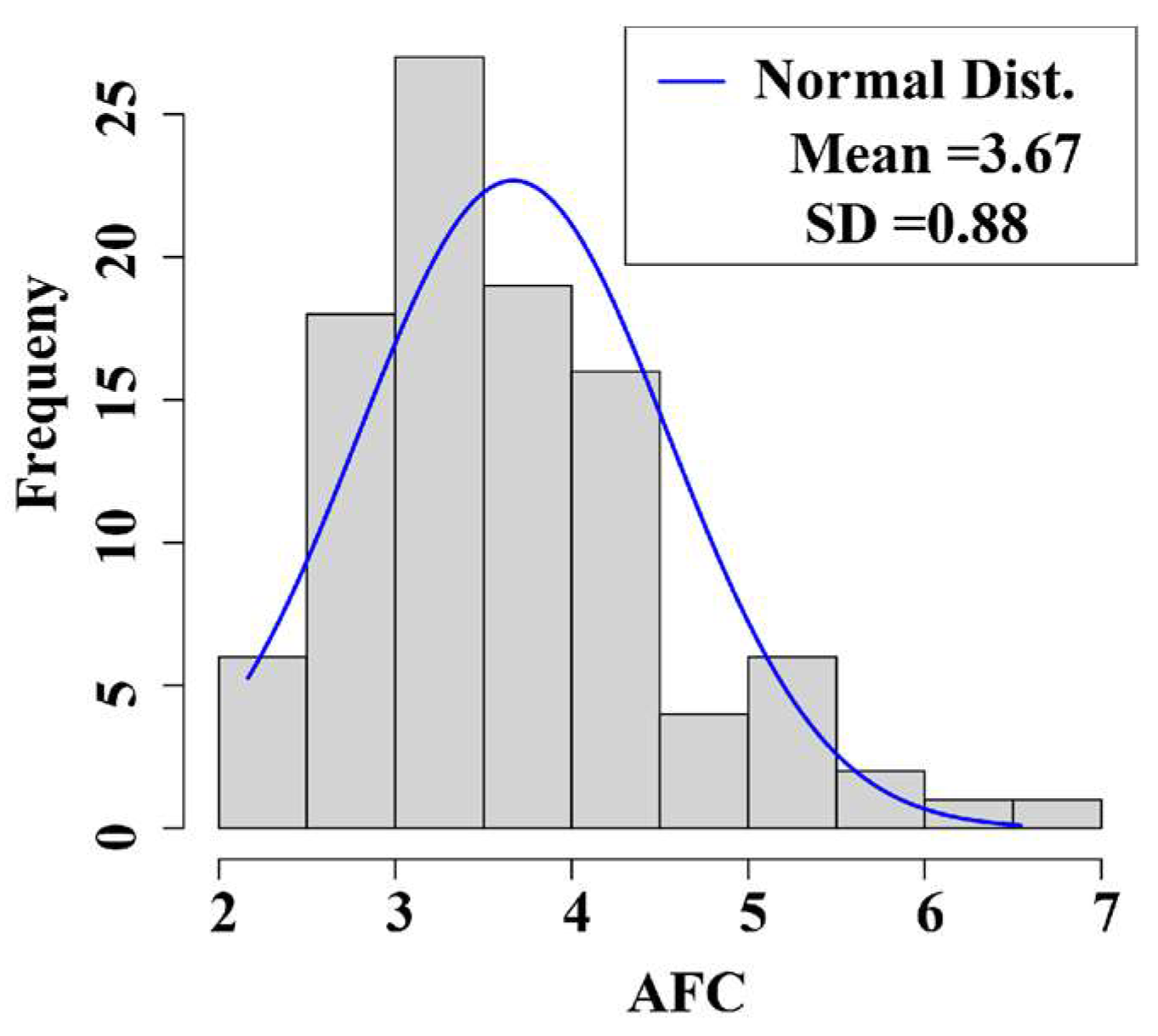
3.1. Phenotypic Value Statistics of the Traits
3.2. Population Structure
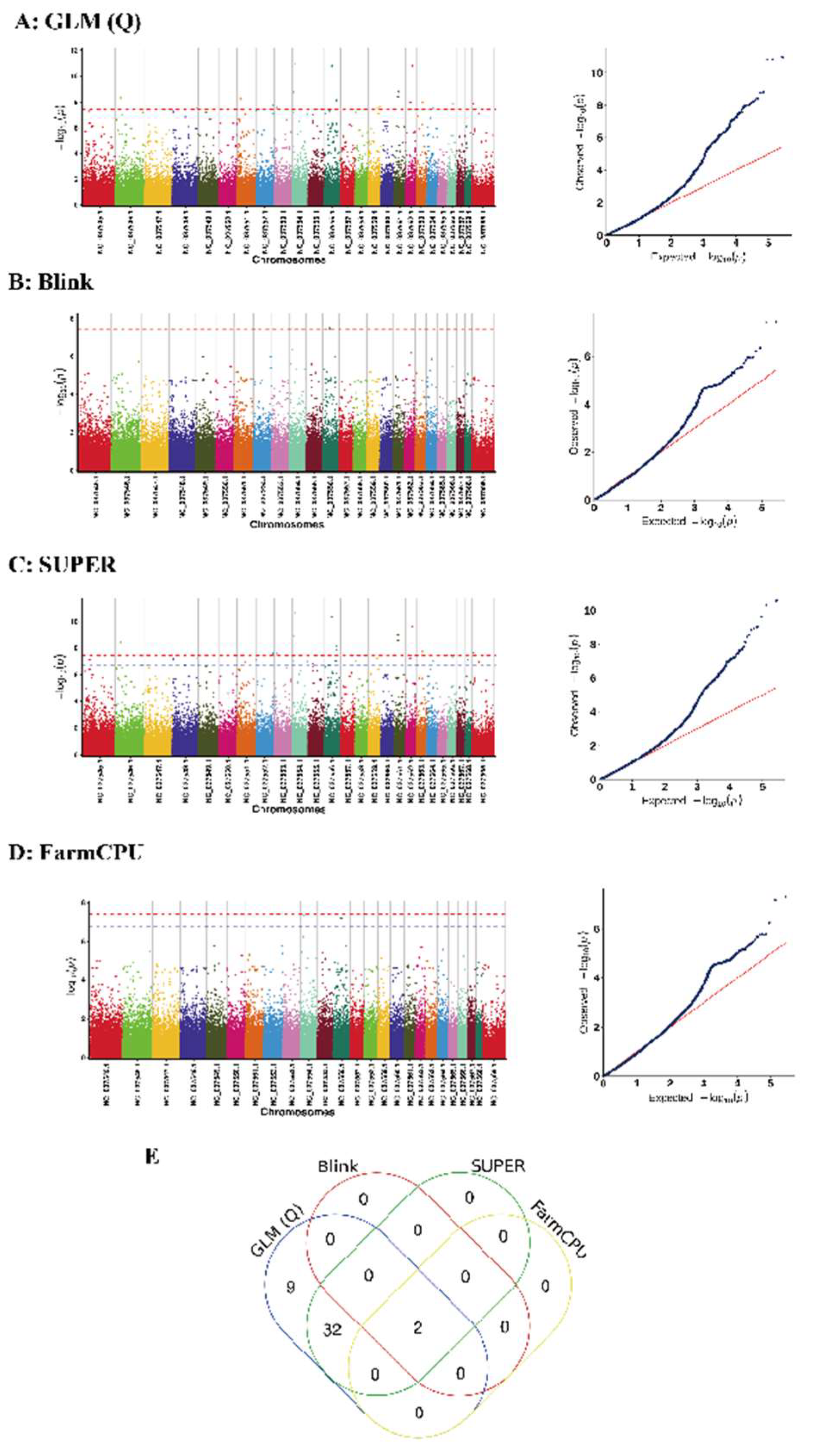
3.3. Results of the Genome-Wide Associations
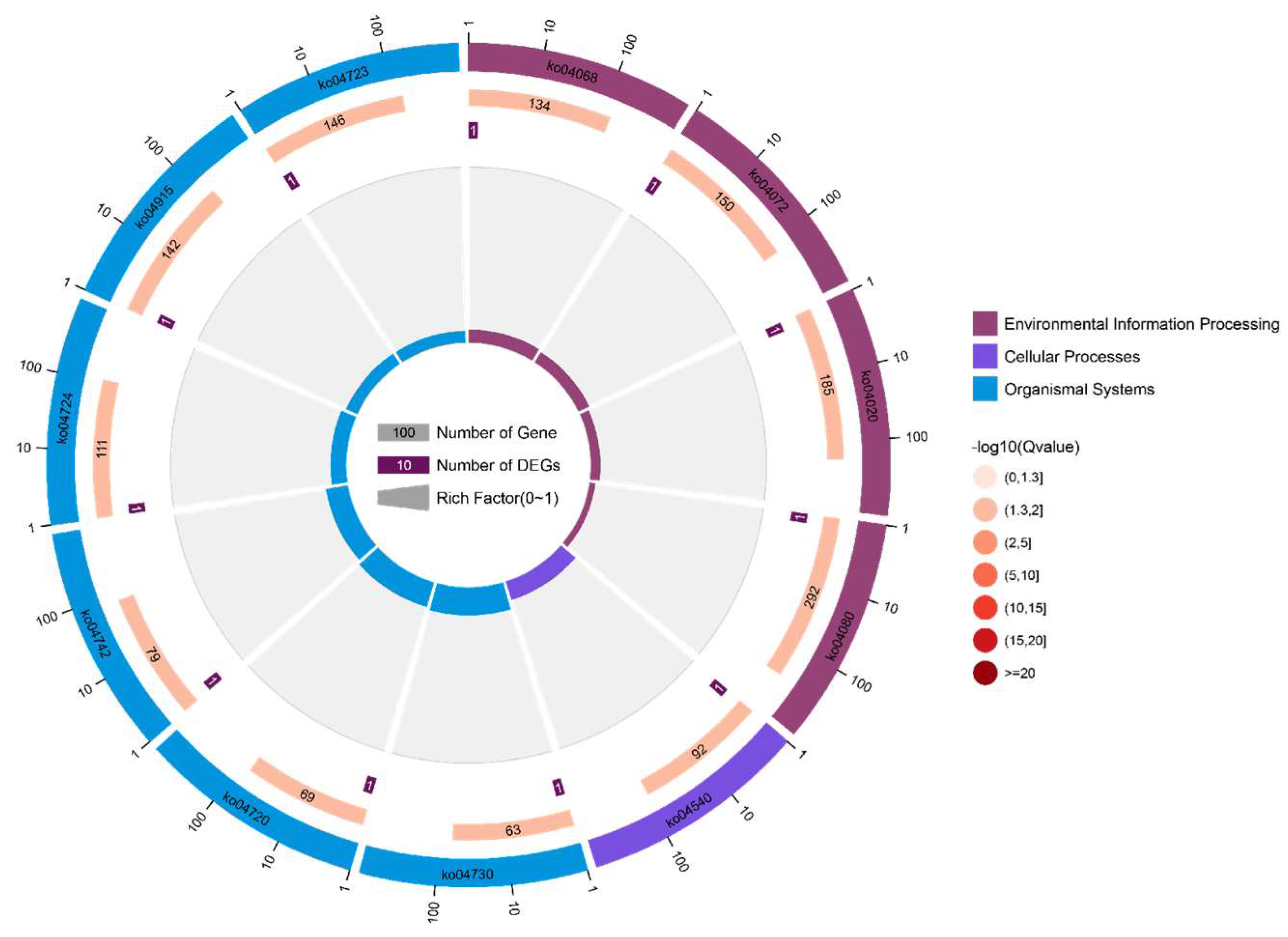
3.4. Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of Candidate Genes
3.5. Significant Association of Fertility with SNP Validation
4. Discussion
4.1. Population Stratification
4.2. Genome-Wide Association Analysis of Reproductive-Related Traits
4.3. The Mechanism of SNP Mutation and Fertility Traits
4.4. Discussion of Tradeoffs
4.5. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Y.X.; Sun, H.; Shaukat, A.; Deng, T.X.; Abdel-Shafy, H.; Che, Z.X.; Zhou, Y.; Hu, C.M.; Li, H.Z.; Wu, Q.P.; et al. Novel Insight into the Role of ACSL1 Gene in Milk Production Traits in Buffalo. Front. Genet. 2022, 13, 896910. [Google Scholar] [CrossRef] [PubMed]
- Barłowska, J.; Szwajkowska, M.; Litwińczuk, Z.; Król, J. Nutritional Value and Technological Suitability of Milk from Various Animal Species Used for Dairy Production. Compr. Rev. Food Sci. Food Saf. 2011, 10, 291–302. [Google Scholar] [CrossRef]
- Brzáková, M.; Čítek, J.; Svitáková, A.; Veselá, Z.; Vostrý, L. Genetic Parameters for Age at First Calving and First Calving Interval of Beef Cattle. Animals 2020, 10, 2122. [Google Scholar] [CrossRef] [PubMed]
- Braganholo, R.C.; Bánkuti, F.I.; dos Santos, F.S.; Pedrosa, V.B.; Zambom, M.A.; Valloto, A.A.; dos Santos, G.T. Age at first calving and productive performance of primiparous Holsteins. J. Dairy Res. 2023, 90, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Srirattana, K.; Hufana-Duran, D.; Atabay, E.P.; Duran, P.G.; Atabay, E.C.; Lu, K.; Liang, Y.; Chaikhun-Marcou, T.; Theerakittayakorn, K.; Parnpai, R. Current status of assisted reproductive technologies in buffaloes. Anim. Sci. J. 2022, 93, e13767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yao, Z.; Li, X.; Zhang, Z.; Liu, X.; Yang, P.; Chen, N.; Xia, X.; Lyu, S.; Shi, Q.; et al. Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data. BMC Genom. 2022, 23, 460. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [PubMed]
- Twomey, A.J.; Cromie, A.R. Impact of age at first calving on performance traits in Irish beef herds. J. Anim. Sci. 2023, 101, skad008. [Google Scholar] [CrossRef]
- Ferrari, V.; Galluzzo, F.; van Kaam, J.B.C.H.M.; Penasa, M.; Marusi, M.; Finocchiaro, R.; Cassandro, M. Genetic and genomic evaluation of age at first calving in Italian Holsteins. J. Dairy Sci. 2024, 107, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Reddy, U.K.; Natarajan, P.; Abburi, V.L.; Bajwa, A.A.; Imran, M.; Zahoor, M.Y.; Abdullah, M.; Bukhari, A.M.; Iqbal, S.; et al. Population demographic history and population structure for Pakistani Nili-Ravi breeding bulls based on SNP genotyping to identify genomic regions associated with male effects for milk yield and body weight. PLoS ONE 2020, 15, e0242500. [Google Scholar] [CrossRef]
- Ye, T.; Shaukat, A.; Yang, L.; Chen, C.; Zhou, Y.; Yang, L. Evolutionary and Association Analysis of Buffalo FABP Family Genes Reveal Their Potential Role in Milk Performance. Genes 2022, 13, 600. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2019, 8, giy154. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Shore, H. Comparison of linear predictors obtained by data transformation, generalized linear models (GLM) and response modeling methodology (RMM). Qual. Reliab. Eng. Int. 2008, 24, 389–399. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, F.; Pan, Y.; Buckler, E.S.; Zhang, Z. A SUPER Powerful Method for Genome Wide Association Study. PLoS ONE 2014, 9, e107684. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.; Tu, Y.; Lv, X. Accelerating BWA-MEM Read Mapping on GPUs. In Proceedings of the 37th International Conference on Supercomputing, Orlando, FL, USA, 21–23 June 2023; Volume 2023, pp. 155–166. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association map** of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Malik, P.L.; Janss, L.; Nielsen, L.K.; Borum, F.; Jørgensen, H.; Eriksen, B.; Rasmussen, S.K. Breeding for dual-purpose wheat varieties using marker–trait associations for biomass yield and quality traits. Theor. Appl. Genet. 2019, 132, 3375–3398. [Google Scholar] [CrossRef] [PubMed]
- Megerssa, S.H.; Sorrells, M.E.; Ammar, K.; Acevedo, M.; Bergstrom, G.C.; Olivera, P.; Abeyo, B. Genome-wide association map** of seedling and adult plant response to stem rust in a durum wheat panel. Plant Genome 2021, 14, e20105. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Liu, X. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Haldar, T.; Ghosh, S. Effect of population stratification on false positive rates of population-based association analyses of quantitative traits. Ann. Hum. Genet. 2012, 76, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Persyn, E.; Redon, R.; Bellanger, L.; Dina, C. The impact of a fine-scale population stratification on rare variant association test results. PLoS ONE 2018, 13, e0207677. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. Principal Component Analyses (PCA)-based findings in population genetic studies are highly biased and must be reevaluated. Sci. Rep. 2022, 12, 14683. [Google Scholar] [CrossRef]
- Nazar, M.; Lu, X.; Abdalla, I.M.; Ullah, N.; Fan, Y.; Chen, Z.; Arbab, A.A.I.; Mao, Y.; Yang, Z. Genome-Wide Association Study Candidate Genes on Mammary System-Related Teat-Shape Conformation Traits in Chinese Holstein Cattle. Genes 2021, 12, 2020. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Zuo, M.; Tong, R.; He, X.; Liu, Y.; Liu, J.; Liu, S.; Liu, Y.; Cao, J.; Ma, L. FOXO Signaling Pathway Participates in Oxidative Stress-Induced Histone Deacetylation. Free Radic. Res. 2023, 57, 47–60. [Google Scholar] [CrossRef]
- Guo, F.; Yu, X.; Xu, A.; Xu, J.; Wang, Q.; Guo, Y.; Kong, L. Japanese encephalitis virus induces apoptosis by inhibiting Foxo signaling pathway. Vet. Microbiol. 2018, 220, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wen, Y.; Jeong, B.S.; Shah, R.; Isola, A.; Silk, A.W.; Goydos, J.S. GRM1 to induce M-CSF and CCL2 expression to lead to an upregulation of M2 macrophages in melanoma. JCO 2017, 35, 122. [Google Scholar] [CrossRef]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent targeting of glutaminolysis and metabotropic glutamate receptor 1 (GRM1) reduces glutamate bioavailability in GRM1+ melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Wall, B.A.; Goydos, J.S.; Chen, S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1). Pigment. Cell Melanoma Res. 2010, 23, 103–111. [Google Scholar] [CrossRef]
- Lange, M.; Begolli, R.; Giakountis, A. Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA 2021, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Hynes, A.C.; Sreenan JM Kane, M.T. The phosphatidylinositol signalling system in elongating bovine blastocysts; formation of phosphoinositides, inositol phosphates and stimulation by growth factors. Reprod. Fertil. Dev. 2002, 14, 515–523. [Google Scholar] [CrossRef] [PubMed]
- López-Gatius, F.; García-Ispierto, I.; Santolaria, P.; Yániz, J.; Nogareda, C.; López-Béjar, M. Screening for high fertility in high-producing dairy cows. Theriogenology 2006, 65, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.A.; Tempelman, R.J.; Allen, M.S.; Weiss, W.P.; Bernard, J.K.; VandeHaar, M.J. Predicting nutrient digestibility in high-producing dairy cows. J. Dairy Sci. 2018, 101, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Reinoso, M.A.; Aponte, P.M.; Cabezas, J.; Rodriguez-Alvarez, L.; Garcia-Herreros, M. Genomic evaluation of primiparous high-producing dairy cows: Inbreeding effects on genotypic and phenotypic production–reproductive traits. Animals 2020, 10, 1704. [Google Scholar] [CrossRef] [PubMed]
- Britt, J.H.; Cushman, R.A.; Dechow, C.D.; Dobson, H.; Humblot, P.; Hutjens, M.F.; Jones, G.A.; Mitloehner, F.M.; Ruegg, P.L.; Sheldon, I.M.; et al. Review: Perspective on high-performing dairy cows and herds. Animal 2021, 15, 100298. [Google Scholar] [CrossRef] [PubMed]
- Nava-Trujillo, H.; Valeris-Chacin, R.; Quintero-Moreno, A.; Escalona-Muñoz, J. Milk yield at first lactation, parity, and season of calving affect the reproductive performance of water buffalo cows. Anim. Prod. Sci. 2020, 60, 1073–1080. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, H.G. Amino acids supplementation for the milk and milk protein production of dairy cows. Animals 2021, 11, 2118. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Somagond, Y.M.; Das, P.; Lathwal, S.S.; Kamboj, A.; Alhussien, M.N.; Dang, A.K. Injection of antioxidant trace minerals/vitamins into peripartum crossbred cows improves the nutritional and immunological properties of colostrum/milk and the health of their calves under heat stress conditions. Trop. Anim. Health Prod. 2024, 56, 225. [Google Scholar] [CrossRef] [PubMed]
- Salles, M.S.; Netto, A.S.; Zanetti, M.A.; Samóra, T.S.; Junior LC, R.; Lima, C.G.; Salles, F.A. Milk biofortification through dietary supplementation of combined selenium, vitamin E and sunflower oil. Livest. Sci. 2022, 258, 104856. [Google Scholar] [CrossRef]
- Strucken, E.M.; Laurenson, Y.C.; Brockmann, G.A. Go with the flow—Biology and genetics of the lactation cycle. Front. Genet. 2015, 6, 118. [Google Scholar] [CrossRef]
- Rodenburg, T.B.; Turner, S.P. The role of breeding and genetics in the welfare of farm animals. Anim. Front. 2012, 2, 16–21. [Google Scholar] [CrossRef]
- Habel, J.; Sundrum, A. Mismatch of glucose allocation between different life functions in the transition period of dairy cows. Animals 2020, 10, 1028. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vicchi, F.; De Winne, C.; Brie, B.; Sorianello, E.; Ladyman, S.R.; Becu-Villalobos, D. Metabolic functions of prolactin: Physiological and pathological aspects. J. Neuroendocrinol. 2020, 32, e12888. [Google Scholar] [CrossRef]
- George, L.; Alex, R.; Sukhija, N.; Jaglan, K.; Vohra, V.; Kumar, R.; Verma, A. Genetic improvement of economic traits in Murrah buffalo using significant SNPs from genome-wide association study. Trop. Anim. Health Prod. 2023, 55, 199. [Google Scholar] [CrossRef]
- Easa, A.A.; El-Aziz, A.H.A.; Barbary, A.S.A.E.; Kostomakhin, N.M.; Nasr, M.A.; Imbabi, T.A. Genetic parameters of production and reproduction traits of Egyptian buffaloes under subtropical conditions. Trop. Anim. Health Prod. 2022, 54, 270. [Google Scholar] [CrossRef] [PubMed]
- Setiaji, A.; Oikawa, T. Genetic parameters of reproductive traits from artificial insemination records of Japanese Black cows. Livest. Sci. 2019, 229, 85–89. [Google Scholar] [CrossRef]
- Castro-Montoya, J.M.; González, F.; Mendoza, M.; Harper, K.; Corea, E.E. Interrelationship between diseases and calving season and their impact on reproductive parameters and milk production of tropical dairy cows. Trop. Anim. Health Prod. 2022, 54, 158. [Google Scholar] [CrossRef]
- Joaquim, L.A.; Changule, A.P.; da Glória Taela, M.; Novela, M.; Pinto, S.C.; Bila, C.G. The generation interval and season of birth do not affect age at first calving, birth weight and calving interval of Mozambican Angoni cattle. Trop. Anim. Health Prod. 2024, 56, 177. [Google Scholar] [CrossRef]
- Hatamian, A.; Levine, L.; Oskouie, H.E.; Sarrafzadeh, M. Exploring a Datasets Statistical Effect Size Impact on Model Performance, and Data Sample-Size Sufficiency. arXiv 2025, arXiv:2501.02673. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Mean | SD | Min | Max |
|---|---|---|---|---|
| AFC | 3.67 | 0.88 | 2.17 | 6.54 |
| Methods | SNP | Chr | Pos | p | R2 | Candidate Genes |
|---|---|---|---|---|---|---|
| SUPER/GLM(Q)/Blink/FarmCPU | 1 | NC_037554.1 | 20,660,384 | 5.0621 × 10−8 | 0.49381 | GRM1 |
| SUPER/GLM(Q)/Blink/FarmCPU | 2 | NC_037556.1 | 50,669,172 | 6.5734 × 10−8 | 0.42667 | -- |
| SUPER/GLM(Q) | 3 | NC_037545.1 | 35,877,289 | 6.8853 × 10−8 | 0.34081 | -- |
| SUPER/GLM(Q) | 4 | NC_037546.1 | 31,601,350 | 3.7412 × 10−9 | 0.41127 | OPN5 |
| SUPER/GLM(Q) | 5 | NC_037548.1 | 4,405,934 | 6.4571 × 10−8 | 0.33847 | FAM118A; UPK3A; KIAA0930 |
| SUPER/GLM(Q) | 6 | NC_037548.1 | 157,620,852 | 5.3121 × 10−8 | 0.3642 | -- |
| SUPER/GLM(Q) | 7 | NC_037551.1 | 20,700,724 | 8.906 × 10−9 | 0.3905 | -- |
| SUPER/GLM(Q) | 8 | NC_037551.1 | 52,947,579 | 1.05 × 10−7 | 0.35825 | -- |
| SUPER/GLM(Q) | 9 | NC_037552.1 | 24,460 | 6.7407 × 10−8 | 0.29687 | -- |
| SUPER/GLM(Q) | 10 | NC_037552.1 | 112,491,377 | 3.2491 × 10−8 | 0.32257 | ZNF777; ZNF746 |
| SUPER/GLM(Q) | 11 | NC_037552.1 | 113,607,118 | 2.4523 × 10−8 | 0.33352 | ABCB8; ASIC3; CDK5; SLC4A2; FASTK; TMUB1; AGAP3 |
| SUPER/GLM(Q) | 12 | NC_037553.1 | 15,992,633 | 2.4361 × 10−8 | 0.36112 | FAM81B |
| SUPER/GLM(Q) | 13 | NC_037554.1 | 9,287,335 | 9.7346 × 10−8 | 0.31436 | TULP4; GTF2H5; SERAC1 |
| SUPER/GLM(Q) | 14 | NC_037554.1 | 16,312,432 | 1.3425 × 10−9 | 0.3879 | -- |
| SUPER/GLM(Q) | 15 | NC_037554.1 | 99,978,913 | 1.0129 × 10−7 | 0.33706 | -- |
| SUPER/GLM(Q) | 16 | NC_037555.1 | 32,169,042 | 1.5181 × 10−7 | 0.33459 | -- |
| SUPER/GLM(Q) | 17 | NC_037556.1 | 75,679,149 | 8.5899 × 10−8 | 0.36278 | -- |
| SUPER/GLM(Q) | 18 | NC_037556.1 | 80,488,833 | 1.413 × 10−8 | 0.39141 | KCNS3; MSGN1 |
| SUPER/GLM(Q) | 19 | NC_037556.1 | 81,118,534 | 7.4243 × 10−9 | 0.36956 | -- |
| SUPER/GLM(Q) | 20 | NC_037558.1 | 8,094,566 | 8.613 × 10−8 | 0.37056 | YBX1; SLC2A10 |
| SUPER/GLM(Q) | 21 | NC_037559.1 | 57,471,045 | 5.8845 × 10−8 | 0.32884 | -- |
| SUPER/GLM(Q) | 22 | NC_037559.1 | 74,176,040 | 3.5625 × 10−8 | 0.35173 | WISP1; NDRG1 |
| SUPER/GLM(Q) | 23 | NC_037561.1 | 25,915,631 | 2.8368 × 10−9 | 0.40653 | -- |
| SUPER/GLM(Q) | 24 | NC_037561.1 | 25,915,784 | 1.0496 × 10−9 | 0.40948 | -- |
| SUPER/GLM(Q) | 25 | NC_037562.1 | 28,221,520 | 6.038 × 10−8 | 0.34565 | -- |
| SUPER/GLM(Q) | 26 | NC_037562.1 | 28,221,526 | 6.038 × 10−8 | 0.34565 | -- |
| SUPER/GLM(Q) | 27 | NC_037562.1 | 28,221,527 | 6.038 × 10−8 | 0.34565 | -- |
| SUPER/GLM(Q) | 28 | NC_037562.1 | 39,920,231 | 2.277 × 10−9 | 0.40894 | HYDIN |
| SUPER/GLM(Q) | 29 | NC_037563.1 | 45,012,914 | 1.8896 × 10−8 | 0.36036 | -- |
| SUPER/GLM(Q) | 30 | NC_037564.1 | 36,725,181 | 9.3193 × 10−8 | 0.31929 | LINGO1 |
| SUPER/GLM(Q) | 31 | NC_037566.1 | 36,784,161 | 4.9686 × 10−8 | 0.3255 | B4GALT6 |
| SUPER/GLM(Q) | 32 | NC_037568.1 | 13,951,865 | 6.9031 × 10−8 | 0.33258 | CALN1 |
| SUPER/GLM(Q) | 33 | NC_037569.1 | 5,832,499 | 2.107 × 10−8 | 0.35009 | KAL1 |
| SUPER/GLM(Q) | 34 | NC_037569.1 | 38,961,581 | 1.0446 × 10−7 | 0.29241 | BCOR |
| Candidate Gene | SNP (Chr:Pos) | Calving Interval (Year) | ||
|---|---|---|---|---|
| Homozygous Mutation | Heterozygous Mutation | Reference Genotype | ||
| GRM1 | NC_037554.1:20660384 | A/A | G/A | G/G |
| 1.02 ± 0.15 B | 1.21 ± 0.23 B | 1.52 ± 0.28 A | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zheng, H.; Cao, D.; Duan, A.; Huang, L.; Feng, C.; Yang, C. GRM1 as a Candidate Gene for Buffalo Fertility: Insights from Genome-Wide Association Studies and Its Role in the FOXO Signaling Pathway. Genes 2025, 16, 193. https://doi.org/10.3390/genes16020193
Li W, Zheng H, Cao D, Duan A, Huang L, Feng C, Yang C. GRM1 as a Candidate Gene for Buffalo Fertility: Insights from Genome-Wide Association Studies and Its Role in the FOXO Signaling Pathway. Genes. 2025; 16(2):193. https://doi.org/10.3390/genes16020193
Chicago/Turabian StyleLi, Wangchang, Haiying Zheng, Duming Cao, Anqin Duan, Liqing Huang, Chao Feng, and Chunyan Yang. 2025. "GRM1 as a Candidate Gene for Buffalo Fertility: Insights from Genome-Wide Association Studies and Its Role in the FOXO Signaling Pathway" Genes 16, no. 2: 193. https://doi.org/10.3390/genes16020193
APA StyleLi, W., Zheng, H., Cao, D., Duan, A., Huang, L., Feng, C., & Yang, C. (2025). GRM1 as a Candidate Gene for Buffalo Fertility: Insights from Genome-Wide Association Studies and Its Role in the FOXO Signaling Pathway. Genes, 16(2), 193. https://doi.org/10.3390/genes16020193