

Genome-Wide Association Study Identifies Potential Regulatory Loci and Pathways Related to Buffalo Reproductive Traits

Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes and Animal Resources
2.2. Sample Collection and Sequencing
2.3. Alignments and Variant Identification
2.4. Variation Filtering
2.5. Principal Component Analysis (PCA)
2.6. Kinship Analysis
2.7. Phylogenetic Tree Construction
2.8. Genome-Wide Association Mapping
3. Results
3.1. Phenotypic Parameters and Marker Information
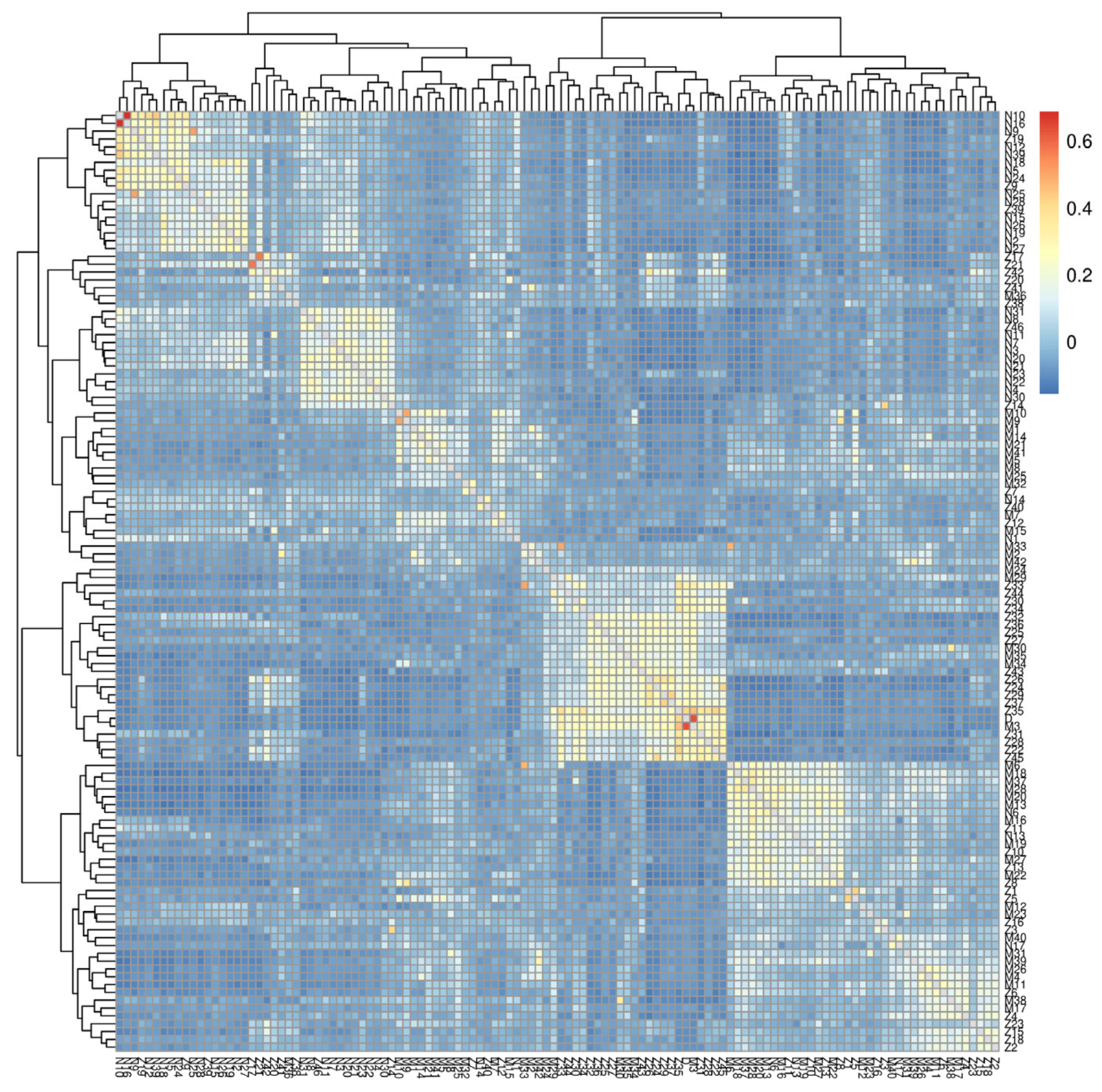
3.2. Population Structure Analysis
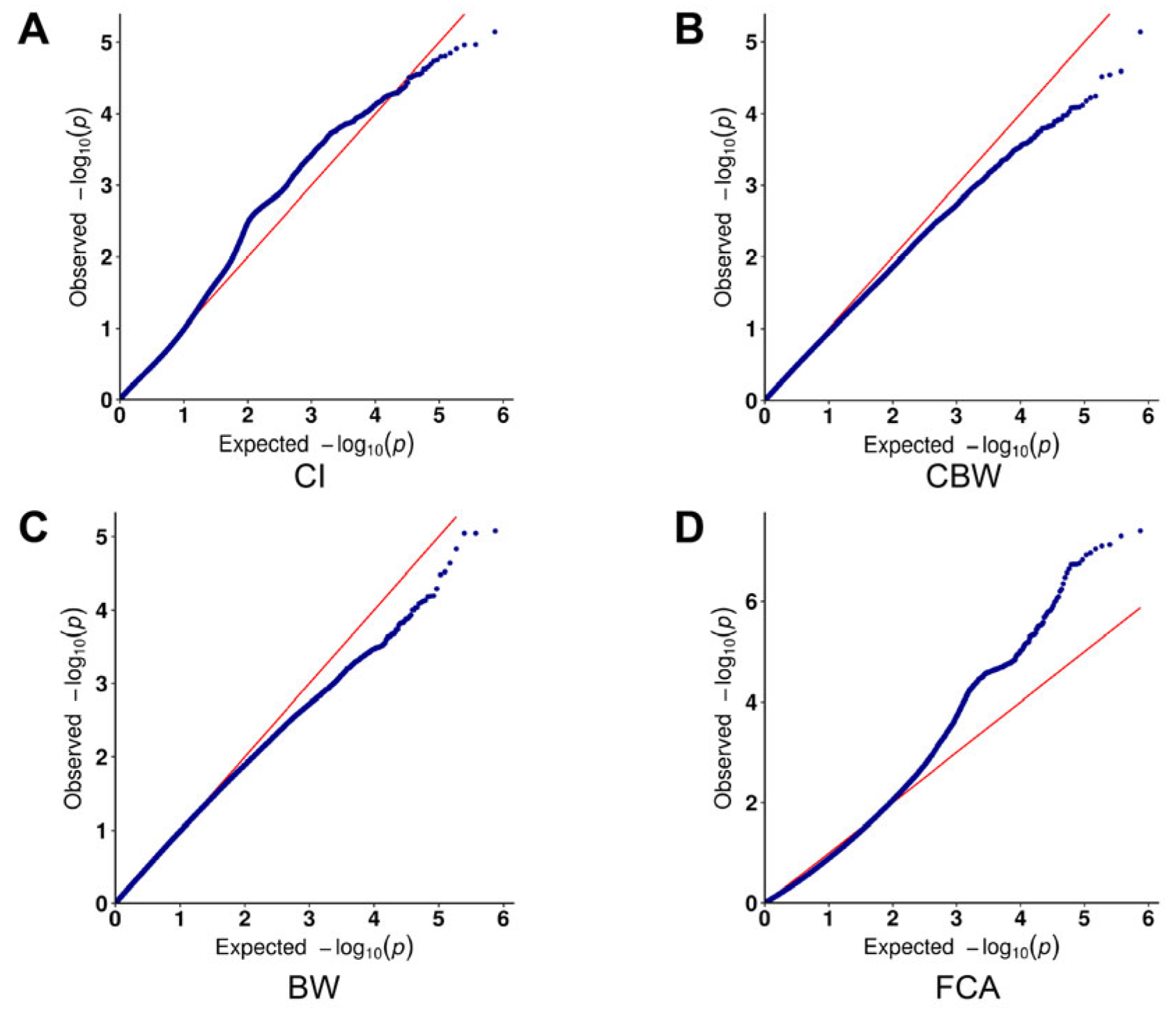
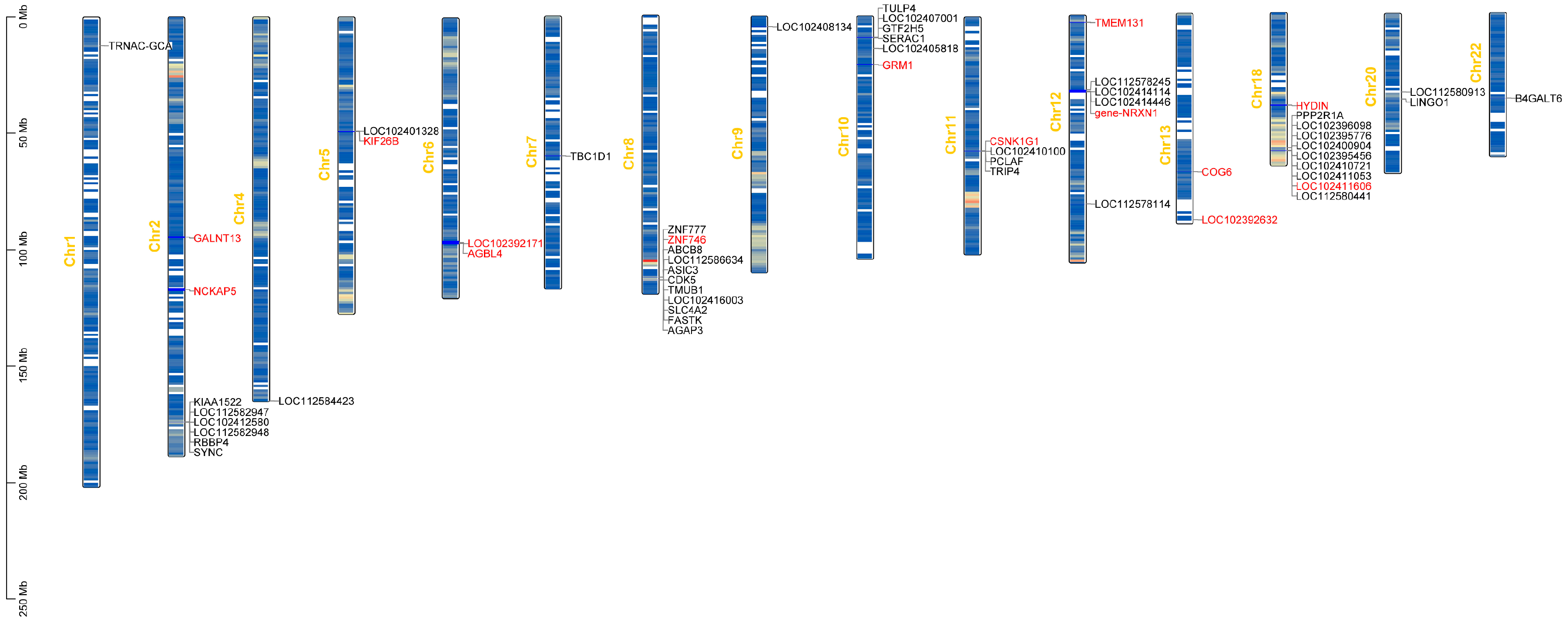
3.3. Results of the Genome-Wide Associations
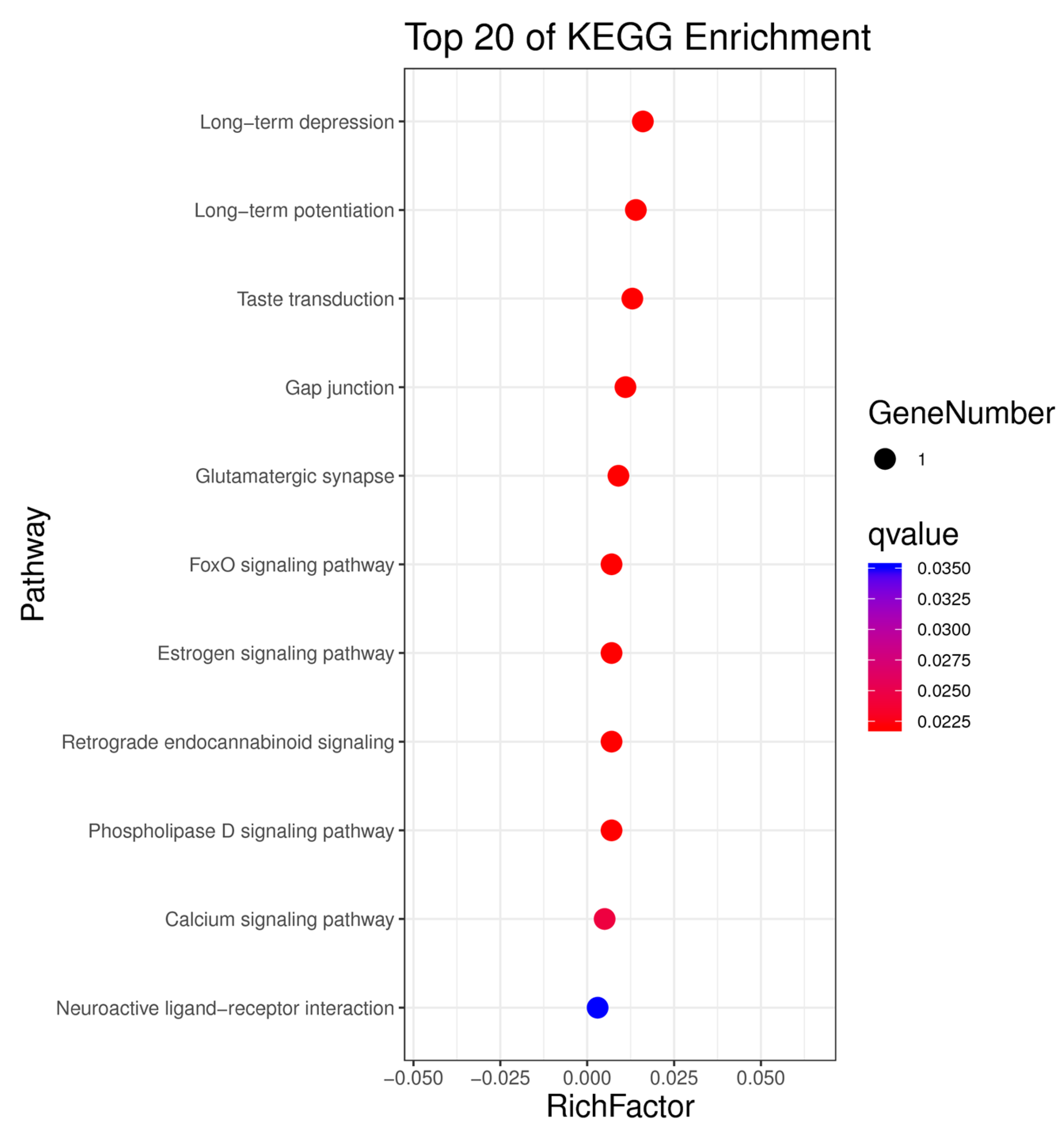
3.4. Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of Candidate Genes
4. Discussion
4.1. Control of Effective Parameters in GWAS Models
4.2. Suggestive Influencing Conditions for Water Buffalo Reproductive Traits and Insights from This Study
4.3. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Khoury, M.J.; Yang, Q. The future of genetic studies of complex human diseases: An epidemiologic perspective. Epidemiology 1998, 9, 350–354. [Google Scholar] [PubMed]
- Van den Berg, I.; Boichard, D.; Lund, M.S. Sequence variants selected from a multi-breed GWAS can improve the reliability of genomic predictions in dairy cattle. Genet. Sel. Evol. 2016, 48, 83. [Google Scholar] [PubMed]
- Wang, Y.; Cao, X.; Luo, C.; Sheng, Z.; Zhang, C.; Bian, C. Multiple ancestral haplotypes harboring regulatory mutations cumulatively contribute to a QTL affecting chicken growth traits. Commun. Biol. 2020, 3, 472. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhou, L.; Huang, Y.; Zheng, M.; Liu, X.; Zhang, Y.; Huang, C.; Peng, S.; Zeng, Q.; Zhong, L.; et al. A whole-genome sequence based association study on pork eating quality traits and cooking loss in a specially designed heterogeneous F6 pig population. Meat Sci. 2018, 146, 160–167. [Google Scholar]
- Zhao, Y.; Hou, Y.; Xu, Y.; Luan, Y.; Zhou, H.; Qi, X.; Hu, M.; Wang, D.; Wang, Z.; Fu, Y.; et al. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat. Commun. 2021, 12, 2217. [Google Scholar]
- Shao, B.; Sun, H.; Ahmad, M.J.; Ghanem, N.; Abdel-Shafy, H.; Du, C.; Deng, T.; Mansoor, S.; Zhou, Y.; Yang, Y.; et al. Genetic features of reproductive traits in bovine and Buffalo: Lessons from bovine to Buffalo. Front. Genet. 2021, 12, 617128. [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- Bovine Genome Sequencing and Analysis Consortium; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522. [Google Scholar]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Schook, L.B. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar]
- Dong, Y.; Xie, M.; Jiang, Y.U.; Xiao, N.; Du, X.; Zhang, W.; Wang, W. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar]
- Huang, Y.; Li, Y.; Burt, D.W.; Chen, H.; Zhang, Y.; Qian, W.; Li, N. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat. Genet. 2013, 45, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Y.; Wang, Z.; Li, X.; Chen, W.; Tao, Z.; Wang, J. The goose genome sequence leads to insights into the evolution of waterfowl and susceptibility to fatty liver. Genome Biol. 2015, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Lee, Y.M.; Ha, J.J.; Jin, S.; Park, B.; Kim, N.Y.; Kim, J.J. Genome-wide association study identifies genomic regions associated with key reproductive traits in Korean Hanwoo cows. BMC Genom. 2024, 25, 496. [Google Scholar] [CrossRef]
- de Fátima Bretanha Rocha, R.; Garcia, A.O.; Otto, P.I.; Santos, M.G.D.; da Silva, M.V.B.; Martins, M.F.; Machado, M.A.; Panetto, J.C.D.C.; Guimarães, S.E.F. Single-step genome-wide association studies and post-GWAS analyses for the number of oocytes and embryos in Gir cattle. Mamm. Genome 2023, 34, 497–508. [Google Scholar] [CrossRef]
- Panigrahi, M.; Rajawat, D.; Nayak, S.S.; Jain, K.; Vaidhya, A.; Prakash, R.; Dutt, T. Genomic insights into key genes and QTLs involved in cattle reproduction. Gene 2024, 917, 148465. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Jiménez, J.M.; Morales, R.M.; Menéndez-Buxadera, A.; Demyda-Peyrás, S.; Laseca, N.; Molina, A. Estimation of the genetic components of (Co) variance and preliminary genome-wide association study for reproductive efficiency in Retinta beef cattle. Animals 2023, 13, 501. [Google Scholar] [CrossRef]
- Li, W.; Zeng, X.; Wang, L.; Yin, L.; Wang, Q.; Yang, H. Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs. Animals 2025, 15, 936. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Stevens, R.C. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 2014, 344, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tourchi, J.J.; Alijani, S.; Rafat, S.A.; Abbasi, M.A. Bayesian-genome-wide association study and post-GWAS on reproductive traits of Holstein dairy cattle. J. Dairy Res. 2024, 91, 146–155. [Google Scholar]
- Mariadassou, M.; Ramayo-Caldas, Y.; Charles, M.; Féménia, M.; Renand, G.; Rocha, D. Detection of selection signatures in Limousin cattle using whole-genome resequencing. Anim. Genet. 2020, 51, 815–819. [Google Scholar] [PubMed]
- Wu, H.Y.; Rong, Y.; Correia, K.; Min, J.; Morgan, J.I. Comparison of the enzymatic and functional properties of three cytosolic carboxypeptidase family members. J. Biol. Chem. 2015, 290, 1222–1232. [Google Scholar]
- Brzáková, M.; Čítek, J.; Svitáková, A.; Veselá, Z.; Vostrý, L. Genetic Parameters for Age at First Calving and First Calving Interval of Beef Cattle. Animals 2020, 10, 2122. [Google Scholar] [CrossRef]
- George, L.; Alex, R.; Sukhija, N.; Jaglan, K.; Vohra, V.; Kumar, R.; Verma, A. Genetic improvement of economic traits in Murrah buffalo using significant SNPs from genome-wide association study. Trop. Anim. Health Prod. 2023, 55, 199. [Google Scholar] [CrossRef]
- Easa, A.A.; El-Aziz, A.H.A.; El Barbary, A.S.A.; Kostomakhin, N.M.; Nasr, M.A.F.; Imbabi, T.A. Genetic parameters of production and reproduction traits of Egyptian buffaloes under subtropical conditions. Trop. Anim. Health Prod. 2022, 54, 270. [Google Scholar]
- Setiaji, A.; Oikawa, T. Genetic parameters of reproductive traits from artificial insemination records of Japanese Black cows. Livest. Sci. 2019, 229, 85–89. [Google Scholar]
- Castro-Montoya, J.M.; González, F.L.; Mendoza, M.V.; Harper, K.; Corea, E.E. Interrelationship between diseases and calving season and their impact on reproductive parameters and milk production of tropical dairy cows. Trop. Anim. Health Prod. 2022, 54, 158. [Google Scholar] [CrossRef]
- Joaquim, L.A.; Changule, A.P.; da Glória Taela, M.; Novela, M.; Pinto, S.C.; Bila, C.G. The generation interval and season of birth do not affect age at first calving, birth weight and calving interval of Mozambican Angoni cattle. Trop. Anim. Health Prod. 2024, 56, 177. [Google Scholar]
- Hatamian, A.; Levine, L.; Oskouie, H.E.; Sarrafzadeh, M. Exploring a Datasets Statistical Effect Size Impact on Model Performance, and Data Sample-Size Sufficiency. arXiv 2025, arXiv:2501.02673. [Google Scholar]
- Sasaki, Y.; Kaneda, S.; Minamino, T.; Honkawa, K.; Horii, Y. Effects of parity, calving season, and previous lactation’s milk yield on fertility of dairy cows on a commercial farm. Anim. Sci. J. 2024, 95, e13998. [Google Scholar] [PubMed]
- Pushpa Magotra, A.; Sindhu, V.; Chaudhary, P. Evaluation of parity and its interaction effect with season of calving, period of calving on production and reproduction traits in Hardhenu (Bos taurus × Bos indicus) dairy cattle. Reprod. Domest. Anim. 2024, 59, e14537. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Records | Mean | SD | Min | Max |
|---|---|---|---|---|---|
| CI | 51 | 581.68 (Days) | 194.21 | 414 | 1336 |
| CBW | 106 | 35.02 (Months) | 4.89 | 18.5 | 46 |
| BW | 115 | 35.47 (Months) | 5.16 | 20 | 46 |
| FCA | 105 | 44.41 (Months) | 15.50 | 5.5 | 120.5 |
| SNP | Chr | Position(bp) | Candidate Gene | p-Value | PVE |
|---|---|---|---|---|---|
| 1 | 1 | 16038140 | TRNAC-GCA | 1.06 × 10−6 | 0.51 |
| 2 | 2 | 94758634 | GALNT13 | 1.54 × 10−6 | 0.50 |
| 3 | 2 | 116842309 | NCKAP5 | 3.08 × 10−6 | 0.37 |
| 4 | 2 | 116842310 | NCKAP5 | 3.08 × 10−6 | 0.37 |
| 5 | 2 | 116842325 | NCKAP5 | 3.08 × 10−6 | 0.37 |
| 6 | 2 | 173941889 | LOC102412580,KIAA1522, SYNC,LOC112582948, RBBP4 | 2.62 × 10−6 | 0.36 |
| 7 | 4 | 164836341 | LOC112584423 | 8.04 × 10−8 | 0.53 |
| 8 | 5 | 49042788 | LOC102401328,KIF26B | 1.37 × 10−6 | 0.36 |
| 9 | 6 | 84593264 | . | 8.09 × 10−7 | 0.55 |
| 10 | 6 | 84593519 | . | 4.45 × 10−7 | 0.54 |
| 11 | 6 | 96791388 | LOC102392171,AGBL4, | 9.2 × 10−8 | 0.51 |
| 12 | 6 | 96811758 | LOC102392171 | 4.69 × 10−6 | 0.33 |
| 12 | 6 | 96811758 | AGBL4 | 4.69 × 10−6 | 0.33 |
| 13 | 7 | 28031213 | . | 5.72 × 10−7 | 0.45 |
| 14 | 7 | 59963243 | TBC1D1 | 8.62 × 10−7 | 0.42 |
| 15 | 7 | 103706036 | . | 2.29 × 10−7 | 0.60 |
| 16 | 8 | 17664369 | . | 2.95 × 10−6 | 0.59 |
| 17 | 8 | 112491377 | ZNF746,ZNF777 | 1.58 × 10−6 | 0.32 |
| 18 | 8 | 113607118 | LOC112586634,ASIC3, LOC102416003,FASTK, TMUB1,ABCB8, CDK5,SLC4A2, AGAP3 | 4.77 × 10−6 | 0.31 |
| 19 | 9 | 4404838 | LOC102408134 | 1.51 × 10−7 | 0.51 |
| 20 | 9 | 4404850 | LOC102408134 | 1.21 × 10−7 | 0.51 |
| 21 | 9 | 4404918 | LOC102408134 | 3.6 × 10−6 | 0.38 |
| 22 | 9 | 4404920 | LOC102408134 | 4.29 × 10−6 | 0.37 |
| 23 | 10 | 9287335 | LOC102407001,GTF2H5, SERAC1 | 2.86 × 10−6 | 0.31 |
| 24 | 10 | 13862680 | LOC102405818 | 1.76 × 10−6 | 0.42 |
| 25 | 10 | 16312432 | . | 6.3 × 10−7 | 0.37 |
| 26 | 10 | 20660384 | GRM1 | 4 × 10−8 | 0.50 |
| 27 | 10 | 45617271 | . | 4.97 × 10−6 | 0.35 |
| 28 | 10 | 45617310 | . | 4.97 × 10−6 | 0.35 |
| 29 | 11 | 32169042 | . | 3.18 × 10−6 | 0.34 |
| 30 | 11 | 57697044 | PCLAF,LOC102410100, TRIP4,CSNK1G1 | 4.69 × 10−6 | 0.33 |
| 31 | 12 | 3176008 | TMEM131 | 2.72 × 10−7 | 0.54 |
| 32 | 12 | 31988278 | LOC112578245,NRXN1 | 1.92 × 10−6 | 0.50 |
| 33 | 12 | 32801224 | LOC102414114,LOC102414446, NRXN1 | 7.54 × 10−8 | 0.54 |
| 34 | 12 | 34019714 | . | 4.38 × 10−6 | 0.38 |
| 35 | 12 | 39801133 | . | 9.78 × 10−7 | 0.61 |
| 36 | 12 | 50669172 | . | 5.07 × 10−8 | 0.43 |
| 37 | 12 | 81013839 | LOC112578114 | 1.11 × 10−7 | 0.49 |
| 38 | 12 | 81118534 | . | 1.49 × 10−6 | 0.35 |
| 39 | 13 | 67984517 | COG6 | 3.56 × 10−6 | 0.42 |
| 40 | 13 | 86238940 | . | 3.38 × 10−7 | 0.54 |
| 41 | 13 | 88603805 | LOC102392632 | 2.9 × 10−6 | 0.40 |
| 42 | 18 | 28221520 | . | 4.57 × 10−6 | 0.37 |
| 43 | 18 | 28221526 | . | 4.57 × 10−6 | 0.37 |
| 44 | 18 | 28221527 | . | 4.57 × 10−6 | 0.37 |
| 45 | 18 | 39920231 | HYDIN | 1.22 × 10−6 | 0.44 |
| 46 | 18 | 58127554 | LOC102396098,LOC102395776, LOC102400904,LOC102395456, PPP2R1A | 1.86 × 10−7 | 0.52 |
| 47 | 18 | 58127556 | LOC102396098,LOC102395776, LOC102400904,LOC102395456, PPP2R1A | 1.86 × 10−7 | 0.52 |
| 48 | 18 | 58127575 | LOC102396098,LOC102395776, LOC102400904,LOC102395456, PPP2R1A | 1.78 × 10−7 | 0.52 |
| 49 | 18 | 58127583 | LOC102396098,LOC102395776, LOC102400904,LOC102395456, PPP2R1A | 1.87 × 10−7 | 0.52 |
| 50 | 18 | 59367852 | LOC112580441,LOC102410721, LOC102411053,LOC102411606 | 2.08 × 10−6 | 0.41 |
| 51 | 20 | 33712610 | LOC112580913 | 1.63 × 10−6 | 0.43 |
| 52 | 20 | 36725181 | LINGO1,B4GALT6 | 2.05 × 10−6 | 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Xie, Q.; Zheng, H.; Duan, A.; Huang, L.; Feng, C.; Shang, J.; Yang, C. Genome-Wide Association Study Identifies Potential Regulatory Loci and Pathways Related to Buffalo Reproductive Traits. Genes 2025, 16, 422. https://doi.org/10.3390/genes16040422
Li W, Xie Q, Zheng H, Duan A, Huang L, Feng C, Shang J, Yang C. Genome-Wide Association Study Identifies Potential Regulatory Loci and Pathways Related to Buffalo Reproductive Traits. Genes. 2025; 16(4):422. https://doi.org/10.3390/genes16040422
Chicago/Turabian StyleLi, Wangchang, Qiyang Xie, Haiying Zheng, Anqin Duan, Liqing Huang, Chao Feng, Jianghua Shang, and Chunyan Yang. 2025. "Genome-Wide Association Study Identifies Potential Regulatory Loci and Pathways Related to Buffalo Reproductive Traits" Genes 16, no. 4: 422. https://doi.org/10.3390/genes16040422
APA StyleLi, W., Xie, Q., Zheng, H., Duan, A., Huang, L., Feng, C., Shang, J., & Yang, C. (2025). Genome-Wide Association Study Identifies Potential Regulatory Loci and Pathways Related to Buffalo Reproductive Traits. Genes, 16(4), 422. https://doi.org/10.3390/genes16040422