
An Actively Homing Insertion Element in a Phage Methylase Contains a Hidden HNH Endonuclease


Abstract
1. Introduction
1.1. Inteins
1.2. SEA-PHAGES and the ShiLan Domain
1.3. The ShiLan Domain Contains an Out-of-Frame HNH Endonuclease
2. Materials and Methods
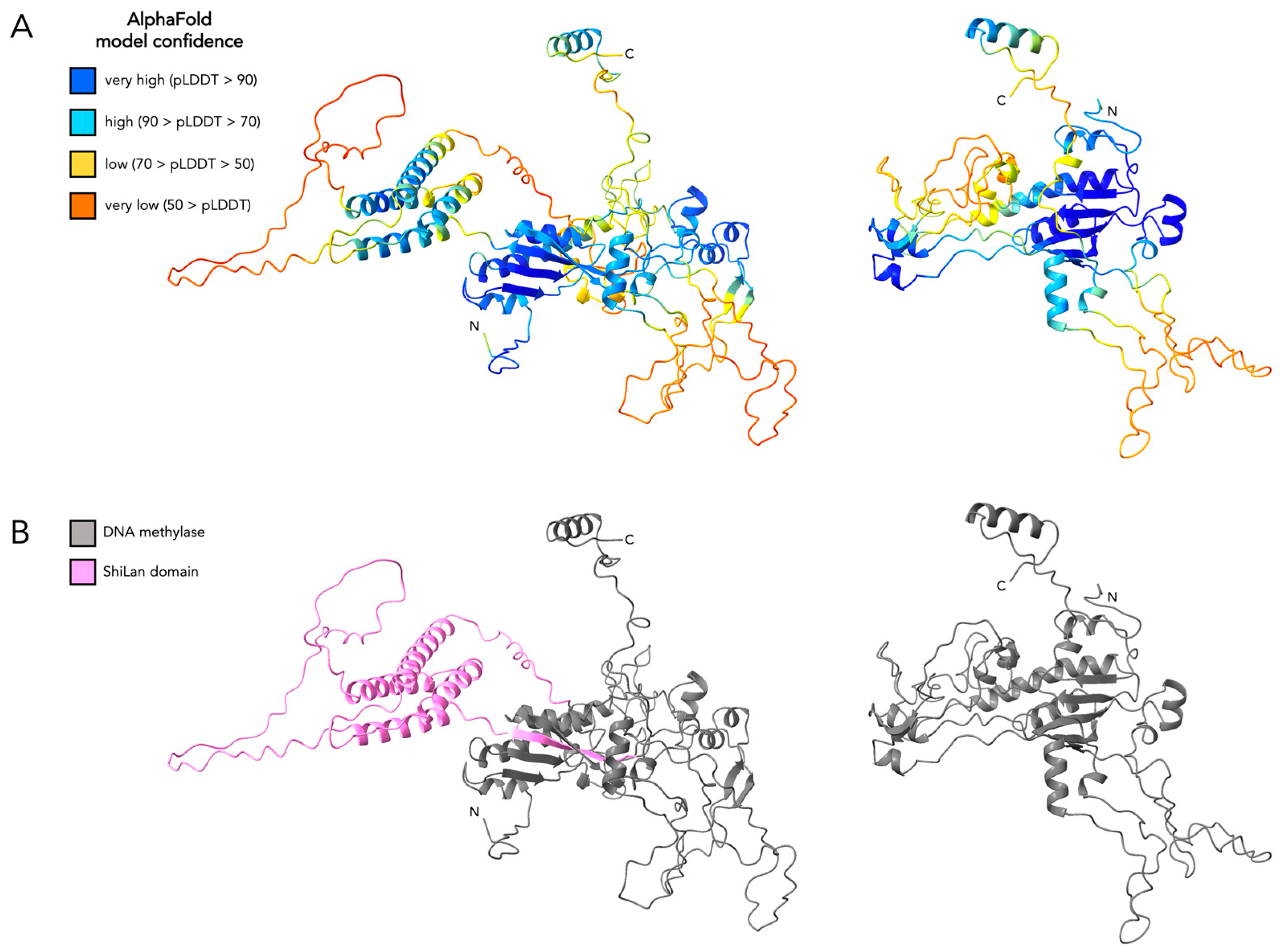
2.1. Protein Structure Prediction and Comparisons
2.2. Databank Searches and Sequence Comparisons
2.3. RNA Secondary Structure Prediction
2.4. Intron Detection with RNAweasel Through MFannot
3. Results
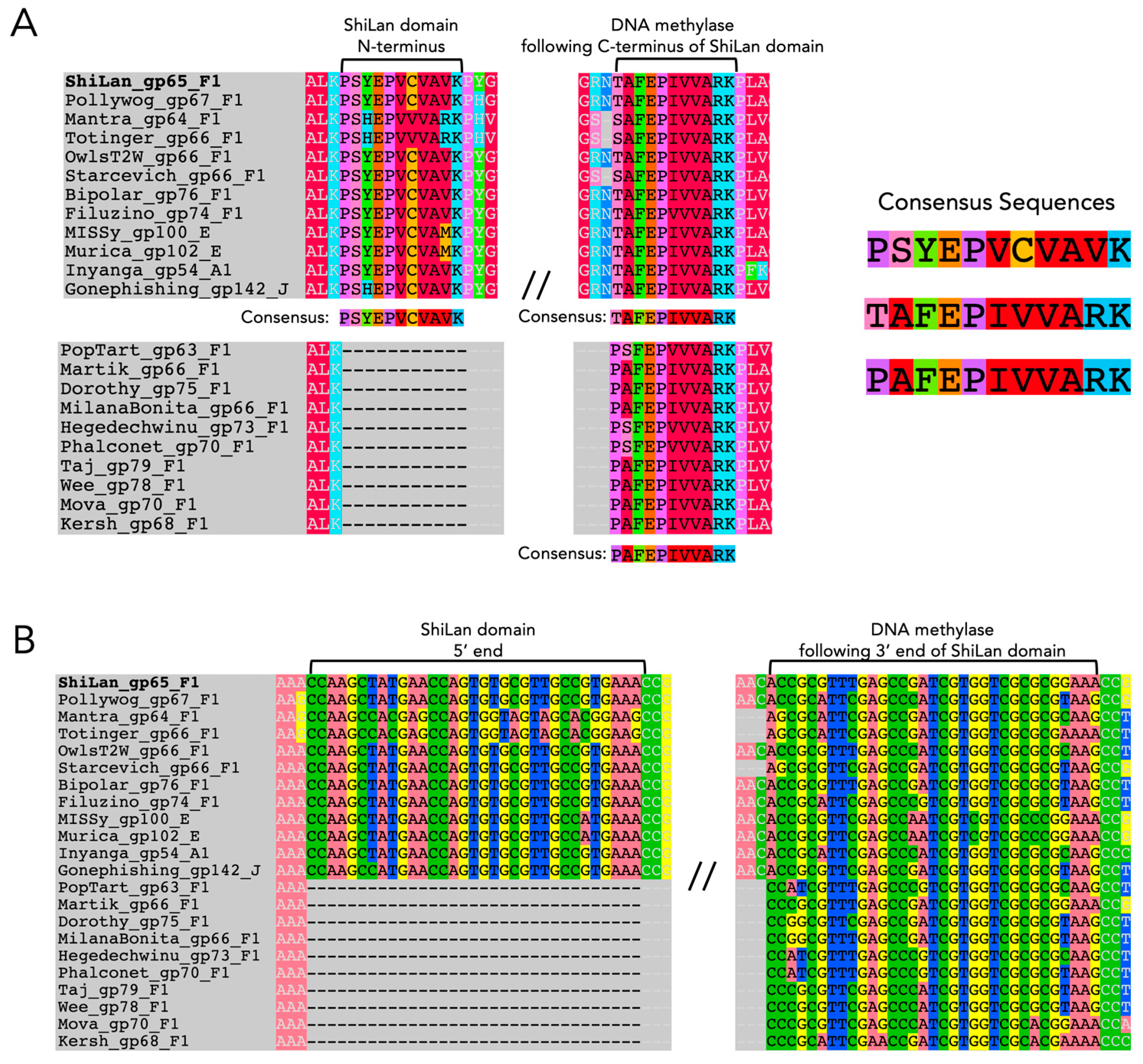
3.1. Assessing the Similarity of ShiLan Domain Nucleotide Sequences to Self-Splicing Introns
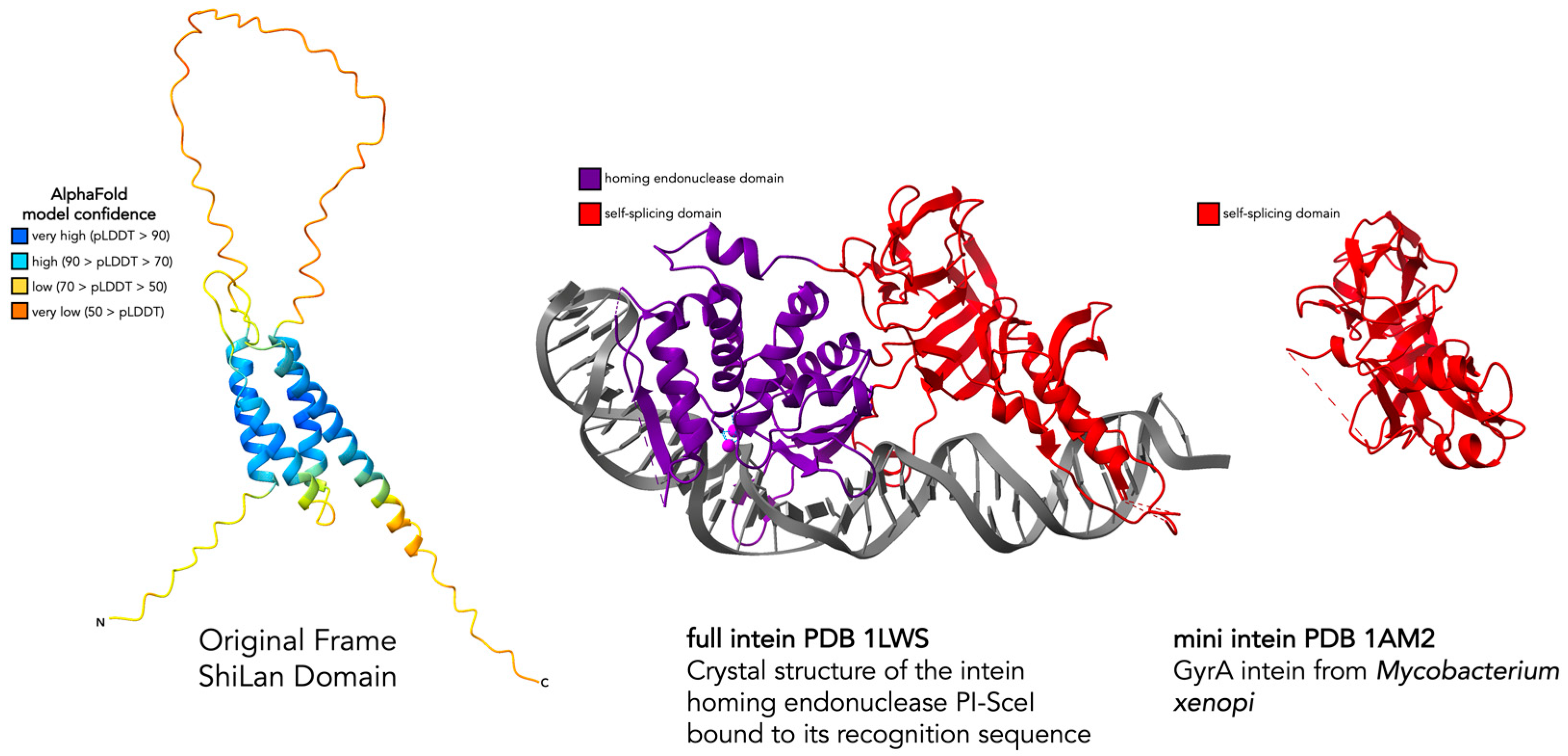
3.2. Assessing the Similarity of ShiLan Domain In-Frame Amino Acid Sequences to Inteins
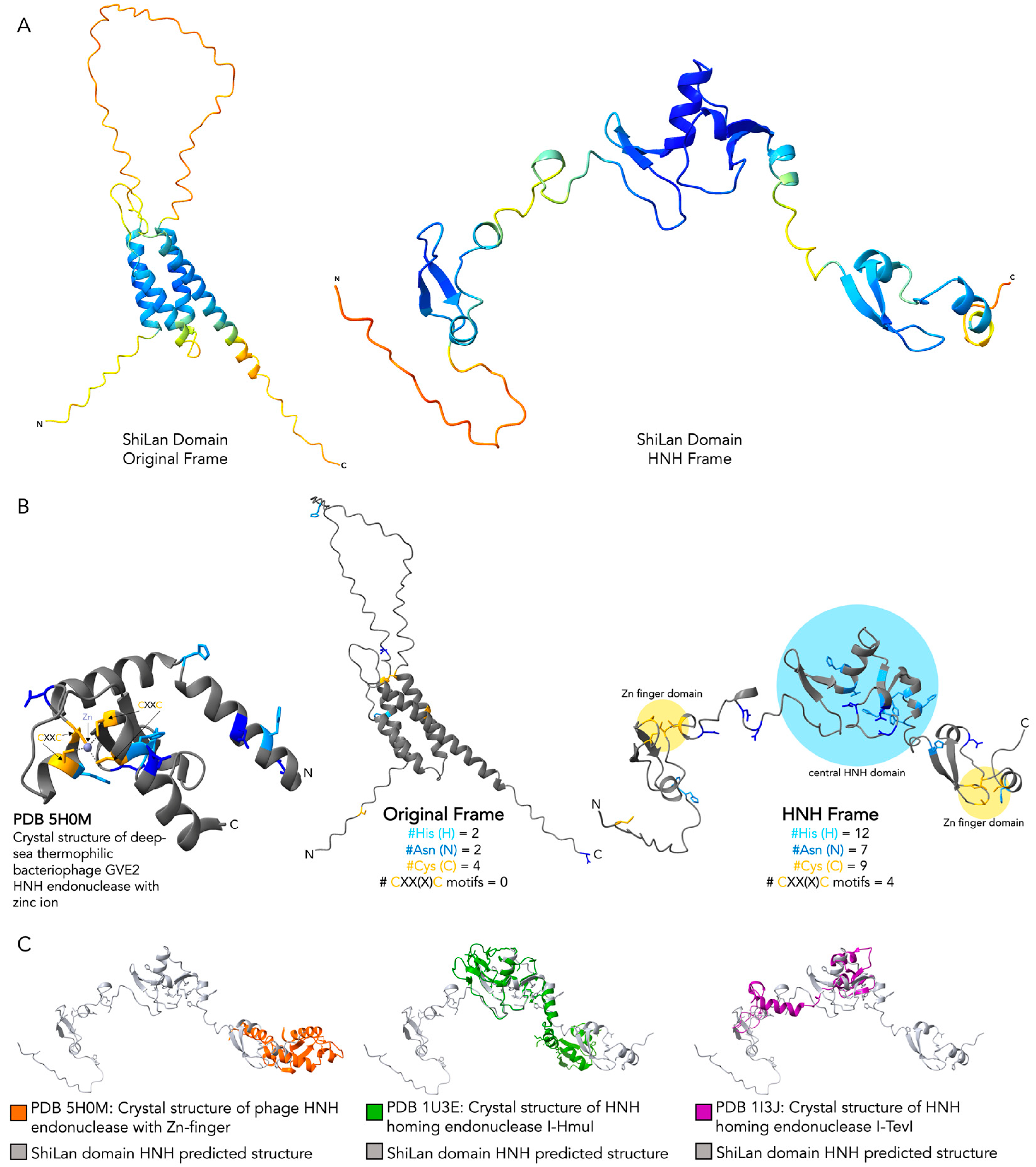
3.3. Comparing Original Frame and HNH Frame ShiLan Domain Protein Products
3.4. Comparing the Predicted Protein Structure of the Original and the HNH Frame Insertion to Solved HNH Endonuclease Structures
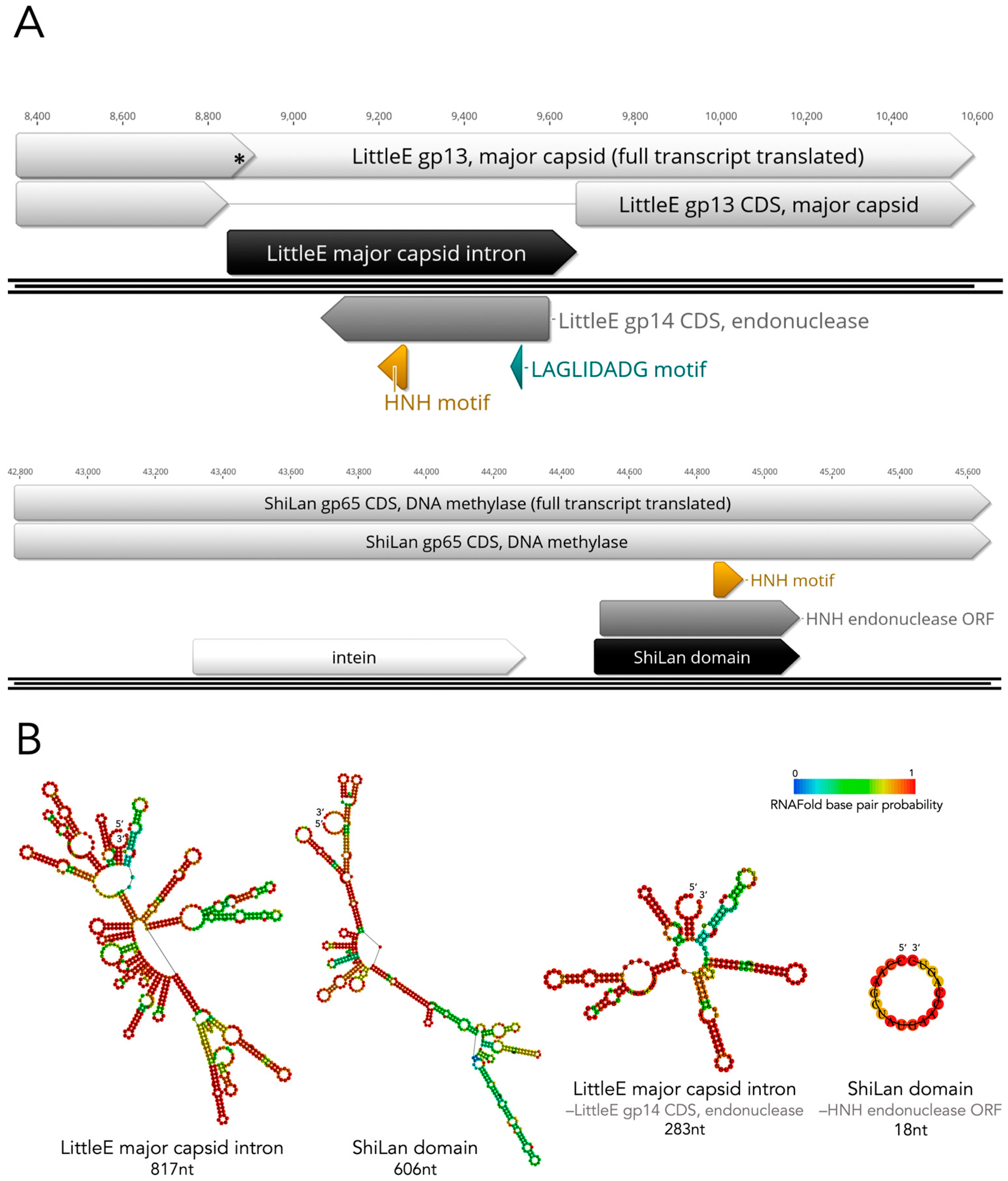
3.5. Revisiting Possible Intron-Based Scenarios in Light of the HNH Discovery
3.6. The Divergent DNA Methylase in Cluster F1 Phage Pacc40 Contains a Different Insertion with a Hidden HNH Endonuclease
4. Discussion
4.1. The ShiLan Domain May Not Interfere with DNA Methylase Function
4.2. DNA Methylases and HNH Endonucleases in Phages
4.3. How Are Hidden HNH Endonucleases Translated?
4.4. Outlook
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid(s) |
| BLASTP | Basic Local Alignment Search Tool for Protein Sequences |
| bp | base pair(s) |
| gp | gene product |
| HNH | type of HE named after a conserved amino acid sequence motif |
| LAGLIDADG | type of HE named after a conserved amino acid sequence motif |
| nt | nucleotide(s) |
| ORF | open reading frame |
| PDB | Protein Data Bank |
| pLDDT | predicted Local Distance Difference Test |
| RBS | ribosomal binding site |
| RM | restriction-modification |
| SEA-PHAGES | Science Education Alliance—Phage Hunters Advancing Genomics and Evolutionary Science |
References
- Soucy, S.M.; Fullmer, M.S.; Papke, R.T.; Gogarten, J.P. Inteins as indicators of gene flow in the halobacteria. Front. Microbiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Kent, S.B.H. Protein splicing: Occurrence, mechanisms and related phenomena. Chem. Biol. 1997, 4, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Perler, F.B.; Xu, M.-Q.; Paulus, H. Protein splicing and autoproteolysis mechanisms. Curr. Opin. Chem. Biol. 1997, 1, 292–299. [Google Scholar] [CrossRef]
- Mills, K.V.; Johnson, M.A.; Perler, F.B. Protein Splicing: How Inteins Escape from Precursor Proteins. J. Biol. Chem. 2014, 289, 14498–14505. [Google Scholar] [CrossRef] [PubMed]
- Novikova, O.; Topilina, N.; Belfort, M. Enigmatic Distribution, Evolution, and Function of Inteins. J. Biol. Chem. 2014, 289, 14490–14497. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, S.P.; Arsenault, D.; Gogarten, J.P. Actinobacteriophage Inteins: Host Diversity, Local Dissemination, and Non-Canonical Architecture. bioRxiv 2025. [Google Scholar] [CrossRef]
- Gosselin, S.P.; Arsenault, D.R.; Jennings, C.A.; Gogarten, J.P. The Evolutionary History of a DNA Methylase Reveals Frequent Horizontal Transfer and Within-Gene Recombination. Genes 2023, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Actinobacteriophages: Genomics, Dynamics, and Applications. Annu. Rev. Virol. 2020, 7, 37–61. [Google Scholar] [CrossRef] [PubMed]
- Tori, K.; Dassa, B.; Johnson, M.A.; Southworth, M.W.; Brace, L.E.; Ishino, Y.; Pietrokovski, S.; Perler, F.B. Splicing of the Mycobacteriophage Bethlehem DnaB Intein. J. Biol. Chem. 2010, 285, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Jacobs-Sera, D.; Best, A.A.; Broussard, G.W.; Connerly, P.L.; Dedrick, R.M.; Kremer, T.A.; Offner, S.; Ogiefo, A.H.; Pizzorno, M.C.; et al. Cluster J Mycobacteriophages: Intron Splicing in Capsid and Tail Genes. PLoS ONE 2013, 8, e69273. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Moure, C.M.; Gimble, F.S.; Quiocho, F.A. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat. Struct. Biol. 2002, 9, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Sharma, S.; Telenti, A.; Jacobs, W.R.; Sacchettini, J.C. Crystal structure of GyrA intein from Mycobacterium xenopi reveals structural basis of protein splicing. Nat. Struct. Mol. Biol. 1998, 5, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, D.; Huang, Y.; Zhu, X.; Rui, M.; Wan, T.; Zheng, X.; Shen, Y.; Chen, X.; Ma, K.; et al. Structural and functional characterization of deep-sea thermophilic bacteriophage GVE2 HNH endonuclease. Sci. Rep. 2017, 7, 42542. [Google Scholar] [CrossRef]
- Shen, B.W.; Landthaler, M.; Shub, D.A.; Stoddard, B.L. DNA Binding and Cleavage by the HNH Homing Endonuclease I-HmuI. J. Mol. Biol. 2004, 342, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, P.; Waddling, C.A.; Fox, K.M.; Belfort, M.; Derbyshire, V. Intertwined structure of the DNA-binding domain of intron endonuclease I-TevI with its substrate. EMBO J. 2001, 20, 3631–3637. [Google Scholar] [CrossRef]
- Shen, B.W.; Heiter, D.F.; Chan, S.-H.; Wang, H.; Xu, S.-Y.; Morgan, R.D.; Wilson, G.G.; Stoddard, B.L. Unusual Target Site Disruption by the Rare-Cutting HNH Restriction Endonuclease PacI. Structure 2010, 18, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2024, 42, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 2021, 3, lqab067. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Geneious Prime v. 2024.0.4.
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A bioinformatic tool for comparative bacteriophage genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Hausner, G.; Hafez, M.; Edgell, D.R. Bacterial group I introns: Mobile RNA catalysts. Mob. DNA 2014, 5, 8. [Google Scholar] [CrossRef]
- Beck, N.; Lang, B.F. RNAweasel, a Webserver for Identification of Mitochondrial, Structured RNAs. 2009. [Online]. Available online: https://github.com/BFL-lab/RNAweasel (accessed on 3 December 2024).
- Beck, N.; Lang, B.F. MFannot, Organelle Genome Annotation Websever. 2010. [Online]. Available online: https://github.com/BFL-lab/Mfannot (accessed on 3 December 2024).
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, R.; Mohr, G.; Belfort, M.; Lambowitz, A.M. Group I and group II introns. FASEB J. 1993, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, D. Supplementary Data and Analyses for ShiLan Domain Update. [Online]. Available online: https://github.com/daniellearsenault/2024_ShiLan_update (accessed on 22 January 2025).
- Turgeman-Grott, I.; Arsenault, D.R.; Yahav, D.; Feng, Y.; Miezner, G.; Naki, D.; Peri, O.; Papke, R.T.; Gogarten, J.P.; Gophna, U. Neighboring inteins interfere with one another’s homing capacity. PNAS Nexus 2023, 2, pgad354. [Google Scholar] [CrossRef]
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef] [PubMed]
- Bonocora, R.P.; Shub, D.A. A Self-Splicing Group I Intron in DNA Polymerase Genes of T7-Like Bacteriophages. J. Bacteriol. 2004, 186, 8153–8155. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Conserved sequences and structures of group I introns: Building an active site for RNA catalysis—A review. Gene 1988, 73, 259–271. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. Methods Mol. Biol. 2021, 2231, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.P.; Rocha, E.P.C.; MacLean, R.C. Restriction-modification systems have shaped the evolution and distribution of plasmids across bacteria. Nucleic Acids Res. 2023, 51, 6806–6818. [Google Scholar] [CrossRef]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; Van Sinderen, D. Bacteriophage Orphan DNA Methyltransferases: Insights from Their Bacterial Origin, Function, and Occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef]
- Leiva, L.E.; Katz, A. Regulation of Leaderless mRNA Translation in Bacteria. Microorganisms 2022, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Cortes, T.; Schubert, O.T.; Rose, G.; Arnvig, K.B.; Comas, I.; Aebersold, R.; Young, D.B. Genome-wide Mapping of Transcriptional Start Sites Defines an Extensive Leaderless Transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013, 5, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.S.; Wang, J.; Lapierre, P.; Mir, M.; Chase, M.R.; Pyle, M.M.; Gawande, R.; Ahmad, R.; Sarracino, D.A.; Ioerger, T.R.; et al. Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLoS Genet. 2015, 11, e1005641. [Google Scholar] [CrossRef] [PubMed]
- Schlub, T.E.; Holmes, E.C. Properties and abundance of overlapping genes in viruses. Virus Evol. 2020, 6, veaa009. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.W.; Molloy, M.P.; Jaschke, P.R. Overlapping genes in natural and engineered genomes. Nat. Rev. Genet. 2022, 23, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E. Non-canonical inteins. Nucleic Acids Res. 1998, 26, 1741–1748. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | ShiLan gp65 (DNA Methylase) ShiLan Domain Insertion Original Frame | ShiLan gp65 (DNA Methylase) ShiLan Domain Insertion HNH Frame |
|---|---|---|
| BLASTP | 9 hits DNA methylase (2) DNA methyltransferase (5) Methyltransferase (2) e-value: 1 × 10−129–2 × 10−58 query cover: 99–100% spans: phages | 100 hits HNH endonuclease (89) Hypothetical protein (11) e-value: 4 × 10−51–1 × 10−12 query cover: 34–86% spans: phages, bacteria, archaea |
| Foldseek (PDB100) | 0 hits with e-value < 1 | 1 hit with e-value < 1 × 10−4 HNH homing endonuclease I-HmuI (PDB entry 1U3E [17]) e-value: 9.13 × 10−3 spans residues 75–173 of query |
| HHpred | 0 hits with e-value < 1 | 51 hits with e-value < 1 × 10−4 HNH endonuclease (38) His-Me/Zn binding (2) endonuclease (7) uncharacterized protein (4) e-value: 8.8 × 10−5–1.4 × 10−13 |
| NCBI Conserved Domain Search | 0 hits with e-value < 1 | 1 hit HNHc superfamily e-value 5.05 × 10−4 spans residues 93–134 of query |
| Program | Pacc40 gp60 (DNA Methylase) Insertion Sequence Original Frame | Pacc40 gp60 (DNA Methylase) Insertion Sequence HNH Frame |
|---|---|---|
| BLASTP | 4 hits DNA methyltransferase (4) query cover: 95–100% spans: phages and bacteria | 100 hits AP2 domain-containing protein (5) HNH endonuclease (2) homing endonuclease (1) hypothetical protein (92) query cover: 87–97% spans: phages and bacteria |
| Foldseek (PDB100) | 0 hits with e-value < 1 | 0 hits with e-value < 1 × 10−4 |
| HHpred | 0 hits with e-value < 1 | 14 hits with e-value < 1 × 10−4 HNH endonuclease (5) restriction endonuclease (1) unknown function (8) e-value: 2.3 × 10−25–2.2 × 10−5 spans residues 4–151 of query |
| NCBI Conserved Domain Search | 0 hits with e-value < 1 | 0 hits with e-value < 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsenault, D.; Gosselin, S.P.; Gogarten, J.P. An Actively Homing Insertion Element in a Phage Methylase Contains a Hidden HNH Endonuclease. Genes 2025, 16, 178. https://doi.org/10.3390/genes16020178
Arsenault D, Gosselin SP, Gogarten JP. An Actively Homing Insertion Element in a Phage Methylase Contains a Hidden HNH Endonuclease. Genes. 2025; 16(2):178. https://doi.org/10.3390/genes16020178
Chicago/Turabian StyleArsenault, Danielle, Sophia P. Gosselin, and Johann Peter Gogarten. 2025. "An Actively Homing Insertion Element in a Phage Methylase Contains a Hidden HNH Endonuclease" Genes 16, no. 2: 178. https://doi.org/10.3390/genes16020178
APA StyleArsenault, D., Gosselin, S. P., & Gogarten, J. P. (2025). An Actively Homing Insertion Element in a Phage Methylase Contains a Hidden HNH Endonuclease. Genes, 16(2), 178. https://doi.org/10.3390/genes16020178