
The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies


 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Methylation Profiling Analysis
2.2. WGS Analysis
2.3. Structural Analysis
3. Results
3.1. Clinical Findings
3.2. Molecular Findings and Structural Analysis
3.3. Assessment of the Clinical Profile Associated with Arg99Gln
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Gillentine, M.A.; Wang, T.; Hoekzema, K.; Rosenfeld, J.; Liu, P.; Guo, H.; Kim, C.N.; De Vries, B.B.A.; Vissers, L.E.L.M.; Nordenskjold, M.; et al. Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 2021, 13, 63–89. [Google Scholar] [CrossRef]
- Shashi, V.; Xie, P.; Schoch, K.; Goldstein, D.B.; Howard, T.D.; Berry, M.N.; Schwartz, C.E.; Cronin, K.; Sliwa, S.; Allen, A.; et al. The RBMX gene as a candidate for the Shashi X-linked intellectual disability syndrome. Clin. Genet. 2015, 88, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M.; Cho, M.T.; Telegrafi, A.; Wilson, A.; Brooks, S.; Botti, C.; Gowans, G.; Autullo, L.A.; Krishnamurthy, V.; Willing, M.C.; et al. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am. J. Hum. Genet. 2016, 99, 728–734. [Google Scholar] [CrossRef]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.L.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Ludecke, H.J.; Hamdan, F.F.; Altmuller, J.; Beleggia, F.; Elcioglu, N.H.; Freyer, C.; Gerkes, E.H.; Demirkol, Y.K.; Knupp, K.G.; et al. Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Hum. Genet. 2017, 136, 821–834. [Google Scholar] [CrossRef]
- Yates, T.M.; Vasudevan, P.C.; Chandler, K.E.; Donnelly, D.E.; Stark, Z.; Sadedin, S.; Willoughby, J.; Broad Center for Mendelian Genomics; DDD Study; Balasubramanian, M. De novo mutations in HNRNPU result in a neurodevelopmental syndrome. Am. J. Med. Genet. 2017, 173, 3003–3012. [Google Scholar] [CrossRef]
- Duijkers, F.A.; McDonald, A.; Janssens, G.E.; Lezzerini, M.; Jongejan, A.; van Koningsbruggen, S.; Leeuwenburgh-Pronk, W.G.; Wlodarski, M.W.; Moutton, S.; Tran-Mau-Them, F.; et al. HNRNPR Variants that Impair Homeobox Gene Expression Drive Developmental Disorders in Humans. Am. J. Hum. Genet. 2019, 104, 1040–1059. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.C.; Li, R.; A Turner, S.; van Jaarsveld, R.H.; Massink, M.P.G.; van den Boogaard, M.J.H.; del Toro, M.; Rodrıguez-Palmero, A.; Fourcade, S.; Schluter, A.; et al. HNRNPH1-related syndromic intellectual disability: Seven additional cases suggestive of a distinct syndromic neurodevelopmental syndrome. Clin. Genet. 2020, 98, 91–98. [Google Scholar] [CrossRef]
- Semino, F.; Schroter, J.; Willemsen, M.H.; Bast, T.; Biskup, S.; Beck-Woedl, S.; Brennenstuhl, H.; Schaaf, C.P.; Kolker, S.; Hoffmann, G.F.; et al. Further evidence for de novo variants in SYNCRIP as the cause of a neurodevelopmental disorder. Hum. Mutat. 2021, 42, 1094–1100. [Google Scholar] [CrossRef]
- Niggl, E.; Bouman, A.; Briere, L.C.; Hoogenboezem, R.M.; Wallaard, I.; Park, J.; Admard, J.; Wilke, M.; Harris-Mostert, E.D.R.O.; Elgersma, M.; et al. HNRNPC haploinsufficiency affects alternative splicing of intellectual disability-associated genes and causes a neurodevelopmental disorder. Am. J. Hum. Genet. 2023, 110, 1414–1435. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Lidéus, S.; Frykhol, C.; Gunnarsson, C.; Mihalic, F.; Gudmundsson, S.; Ekvall, S.; Molin, A.M.; Pham, M.; Vihinen, M.; et al. Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions. Eur. J. Hum. Genet. 2024, 32, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Herzner, A.M.; Khan, Z.; van Nostrand, E.L.; Chan, S.; Cuellar, T.; Chen, R.; Pechuan-Jorge, X.; Komuves, L.; Solon, M.; Modrusan, Z.; et al. ADAR and hnRNPC deficiency synergize in activating endogenous dsRNA-induced type I IFN responses. J. Exp. Med. 2021, 218, e20201833. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, A.; Taniguchi, I.; Shinmyozu, K.; Ohno, M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 2012, 335, 1643–1646. [Google Scholar] [CrossRef] [PubMed]
- Görlach, M.; Burd, C.G.; Dreyfuss, G. The determinants of RNA-binding specificity of the heterogeneous nuclear ribonucleoprotein C proteins. J. Biol. Chem. 1994, 269, 23074–23078. [Google Scholar] [CrossRef] [PubMed]
- Koloteva-Levine, N.; Amichay, M.; Elroy-Stein, O. Interaction of hnRNP-C1/C2 proteins with RNA: Analysis using the yeast three-hybrid system. FEBS Lett. 2002, 523, 73–78. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Verloes, A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am. J. Med. Genet. A 2005, 133, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.D.; Prasad, C. CHARGE syndrome. Orphanet. J. Rare Dis. 2006, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Ciolfi, A.; Foroutan, A.; Capuano, A.; Pedace, L.; Travaglini, L.; Pizzi, S.; Andreani, M.; Miele, E.; Invernizzi, F.; Reale, C.; et al. Childhood-onset dystonia-causing KMT2B variants result in a distinctive genomic hypermethylation profile. Clin. Epigenet. 2021, 13, 157. [Google Scholar] [CrossRef] [PubMed]
- Niceta, M.; Ciolfi, A.; Ferilli, M.; Pedace, L.; Cappelletti, C.; Nardini, C.; Hildonen, M.; Chiriatti, L.; Miele, E.; Dentici, M.L.; et al. DNA methylation profiling in Kabuki syndrome: Reclassification of germline KMT2D VUS and sensitivity in validating postzygotic mosaicism. Eur. J. Hum. Genet. 2024, 32, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Cytrynbaum, C.; Turinsky, A.L.; Siu, M.T.; Inbar-Feigenberg, M.; Mendoza-Londono, R.; Chitayat, D.; Walker, S.; Machado, J.; Caluseriu, O.; et al. CHARGE and Kabuki syndromes: Gene-specific DNA Methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet. 2017, 100, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Groupe DI France; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Ferilli, M.; Ciolfi, A.; Pedace, L.; Niceta, M.; Radio, F.C.; Pizzi, S.; Miele, E.; Cappelletti, C.; Mancini, C.; Galluccio, T.; et al. Genome-Wide DNA Methylation Profiling Solves Uncertainty in Classifying NSD1 Variants. Genes 2022, 13, 2163. [Google Scholar] [CrossRef]
- Motta, M.; Fasano, G.; Gredy, S.; Brinkmann, J.; Bonnard, A.A.; Simsek-Kiper, P.O.; Gulec, E.Y.; Essaddam, L.; Utine, G.E.; Prandi, I.G.; et al. SPRED2 loss-of-function causes a recessive Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2021, 108, 2112–2129. [Google Scholar] [CrossRef] [PubMed]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Köhler, S.; Zemojtel, T.; Spielmann, M.; Jäger, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, 333–339. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for Structural Variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Cieniková, Z.; Damberger, F.F.; Hall, J.; Allain, F.H.; Maris, C. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J. Am. Chem. Soc. 2014, 136, 14536–14544. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Safiullah, A.M.; Fernbach, S.D.; Harutyunyan, K.G.; Thaller, C.; Peterson, L.E.; McPherson, J.D.; Gibbs, R.A.; White, L.D.; Hefner, M.; et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 2006, 78, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.A.; Juliano, A.; Robson, C.; Clement, E.; Nash, R.; Rajput, K.; D’Arco, F. The spectrum of cochlear malformations in CHARGE syndrome and insights into the role of the CHD7 gene during embryogenesis of the inner ear. Neuroradiology 2023, 65, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Szleper, A.; Lachowska, M.; Wojciechowski, T.; Pronicka-Iwanicka, K. Detailed analysis of inner ear malformations in CHARGE syndrome patients—Correlation with audiological results and proposal for computed tomography scans evaluation methodology. Braz. J. Otorhinolaryngol. 2024, 90, 101383. [Google Scholar] [CrossRef] [PubMed]
- Janssen, N.; Bergman, J.E.; Swertz, M.A.; Tranebjaerg, L.; Lodahl, M.; Schoots, J.; Hofstra, R.M.; van Ravenswaaij-Arts, C.M.; Hoefsloot, L.H. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum. Mutat. 2012, 33, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Moccia, A.; Srivastava, A.; Skidmore, J.M.; Bernat, J.A.; Wheeler, M.; Chong, J.X.; Nickerson, D.; Bamshad, M.; Hefner, M.A.; Martin, D.M.; et al. Genetic analysis of CHARGE syndrome identifies overlapping molecular biology. Genet. Med. 2018, 20, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.Ö.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef]
- Baldridge, D.; Spillmann, R.C.; Wegner, D.J.; Wambach, J.A.; White, F.V.; Sisco, K.; Toler, T.L.; Dickson, P.I.; Cole, F.S.; Shashi, V.; et al. Phenotypic expansion of KMT2D-related disorder: Beyond Kabuki syndrome. Am. J. Med. Genet. A 2020, 182, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Cuvertino, S.; Hartill, V.; Colyer, A.; Garner, T.; Nair, N.; Al-Gazali, L.; Canham, N.; Faundes, V.; Flinter, F.; Hertecant, J.; et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet. Med. 2020, 22, 867–877. [Google Scholar] [CrossRef]
- Stadelmaier, R.T.; Kenna, M.A.; Barrett, D.; Mullen, T.E.; Bodamer, O.; Agrawal, P.B.; Robson, C.D.; Wojcik, M.H. Neuroimaging in Kabuki syndrome and another KMT2D-related disorder. Am. J. Med. Genet. A 2021, 185, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Fregeau, B.; Kim, B.J.; Hernández-García, A.; Jordan, V.K.; Cho, M.T.; Schnur, R.E.; Monaghan, K.G.; Juusola, J.; Rosenfeld, J.A.; Bhoj, E.; et al. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am. J. Hum. Genet. 2016, 98, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.K.; Fregeau, B.; Ge, X.; Giordano, J.; Wapner, R.J.; Balci, T.B.; Carter, M.T.; Bernat, J.A.; Moccia, A.N.; Srivastava, A. Genotype-phenotype correlations in individuals with pathogenic RERE variants. Hum. Mutat. 2018, 39, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Menke, L.A.; DDD Study; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 2018, 176, 862–876. [Google Scholar] [CrossRef]

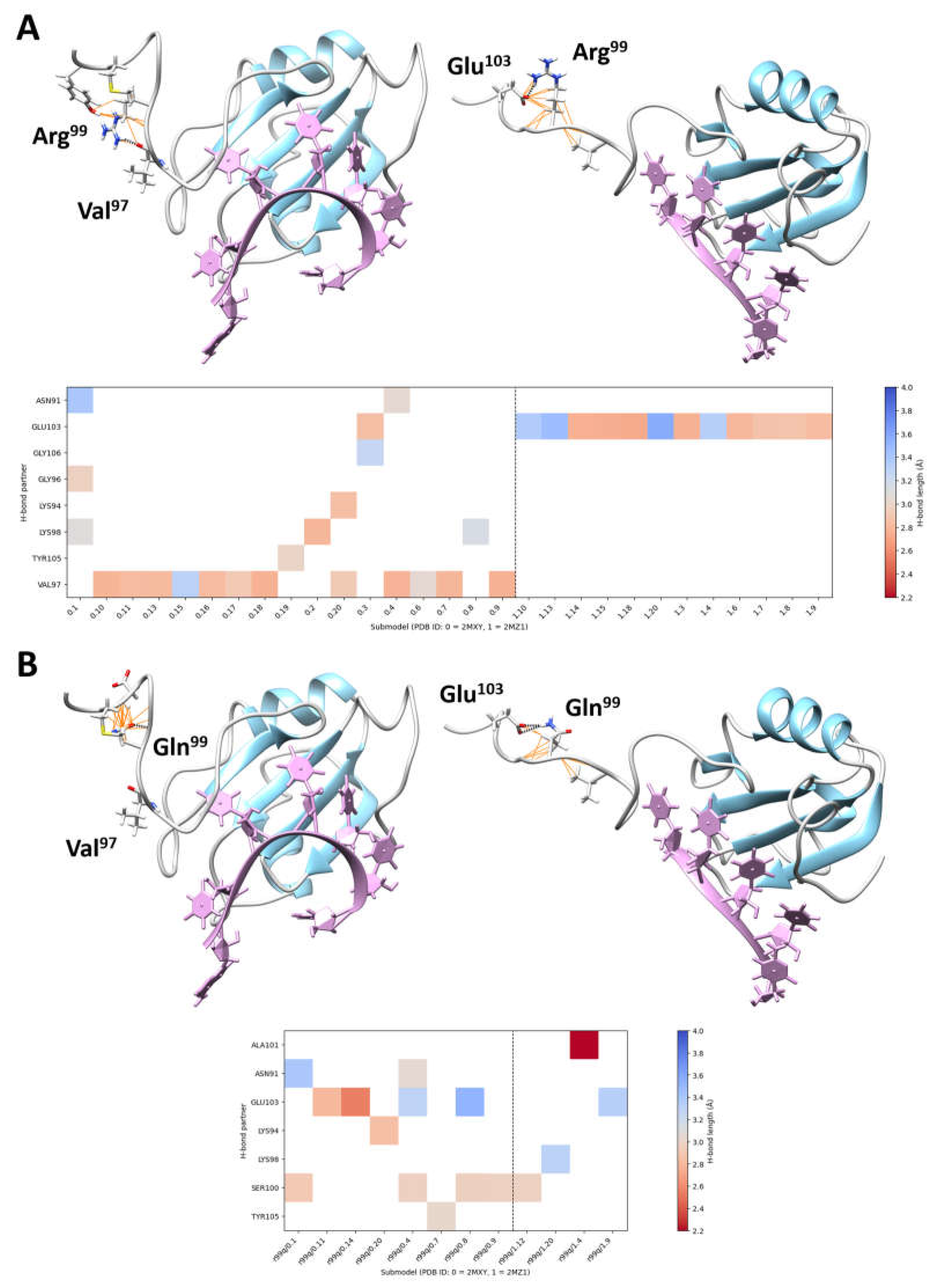

{kind=link}
{kind=link}
| Present Study | Individual 11 [11] | |
|---|---|---|
| Sex | Female | Female |
| Age at evaluation | 14 years | 4 years 5 months |
| HNRNPC variant | c.296G>A, p.Arg99Gln (de novo) | c.296G>A, p.Arg99Gln (de novo) |
| ACMG variant classification | PS2, PM2 (likely pathogenic) | PS2, PM2 (likely pathogenic) |
| Molecular analysis | WGS (trio) | WGS (trio) |
| Other clinically relevant variants | None | None |
| Phenotypic information | ||
| Pregnancy and birth | Normal | Normal, urinary tract infection |
| Gestational age (weeks) | 40 | 40 + 4 |
| Weight at birth (g) | 3730 | 3540 |
| Length at birth (cm) | 51 | |
| OFC at birth (cm) | 36 | 33 |
| Apgar score | 8–9 | |
| Growth parameters | 18 years | 6 years, 8 months |
| Height (cm) | 142.3 cm (−3.0 SD); | 122.9 cm (0.7 SD) |
| Weight (kg) | 40.9 kg (−1.8 SD); | 29.5 kg (1.7 SD) |
| OFC (cm) | 50 cm (−3.0 SD) | 4y5m: 49.3 cm (−1.7 SD) |
| First words | No words | 3 years |
| Walking | 2.5 years | 2 years |
| Language development | No words | Few single words and signs |
| Gross motor skills | Mild delay | Mild delay |
| Cognitive deficit | Moderate (IQ 42) | Mild |
| Seizures | No | No |
| Hypo-/hypertonia | No | No |
| Movement disorder | No | No |
| MRI anomalies | Aplasia of left cochlea and hypoplasia of the right cochlea | Absent cochlea (right), hypoplasia of the left cochlea, absent cochlear nerves |
| Behavioral anomalies | Stress-related hetero-aggressiveness, stereotypies | None |
| Sleeping problems | No | No |
| Facial dysmorphism | Broad and slightly receding forehead, high upper hairline, thick eyebrows, mild synophrys, wave-shaped lids, mild eversion of lower lateral eyelids, narrow and upslanted palpebral fissures, mildly deep-set eyes, deep and long philtrum, sharp nose, anteverted nares, thin upper lip, prominent malar region, preauricular fistula | Broad and slightly receding forehead, high upper hairline, mildly thick eyebrows, mild synophrys, wave-shaped lids, everted lower lateral eyelids, mildly upslanted palpebral fissures, deep-set eyes, smooth and long philtrum, anteverted nares, thin upper lip, prominent malar region, notched lower lateral incisor |
| Eyes/vision | Bilateral colobomatous microphthalmia | Bilateral colobomatous microphthalmia |
| Hearing | Sensorineural hearing loss | Sensorineural hearing loss |
| Cardiac problems | Patent ductus arteriosus | |
| Renal problems | None | None |
| Endocrinological problems | Delayed menarche, growth delay, pituitary gland hypoplasia | Premature thelarche |
| Additional features | Short and broad distal phalanx of the first finger of the hands, hirsutism | Prominent finger pads, hirsute legs, single gray scalp hair |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiriatti, L.; Priolo, M.; Onesimo, R.; Carvetta, M.; Leoni, C.; Bruselles, A.; Radio, F.C.; Cappelletti, C.; Ferilli, M.; Ricci, D.; et al. The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies. Genes 2025, 16, 176. https://doi.org/10.3390/genes16020176
Chiriatti L, Priolo M, Onesimo R, Carvetta M, Leoni C, Bruselles A, Radio FC, Cappelletti C, Ferilli M, Ricci D, et al. The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies. Genes. 2025; 16(2):176. https://doi.org/10.3390/genes16020176
Chicago/Turabian StyleChiriatti, Luigi, Manuela Priolo, Roberta Onesimo, Mattia Carvetta, Chiara Leoni, Alessandro Bruselles, Francesca Clementina Radio, Camilla Cappelletti, Marco Ferilli, Daniela Ricci, and et al. 2025. "The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies" Genes 16, no. 2: 176. https://doi.org/10.3390/genes16020176
APA StyleChiriatti, L., Priolo, M., Onesimo, R., Carvetta, M., Leoni, C., Bruselles, A., Radio, F. C., Cappelletti, C., Ferilli, M., Ricci, D., Niceta, M., Cordeddu, V., Ciolfi, A., Mancini, C., Zampino, G., & Tartaglia, M. (2025). The Arg99Gln Substitution in HNRNPC Is Associated with a Distinctive Clinical Phenotype Characterized by Facial Dysmorphism and Ocular and Cochlear Anomalies. Genes, 16(2), 176. https://doi.org/10.3390/genes16020176