Abstract
Otofaciocervical syndrome (OTFCS) is a rare disorder characterized by facial, auditory, and shoulder girdle anomalies. Its significant phenotypic overlap with branchiootorenal spectrum disorders (BORSD)—both linked to EYA1 (EYA transcriptional coactivator and phosphatase 1) gene defects—has raised questions about whether they are distinct entities or part of a single clinical spectrum. We report a novel OTFCS patient with a de novo microdeletion spanning EYA1 and review all published cases of EYA1-related disorders. Our analysis reveals that all EYA1 variant types (truncating, missense, CNV, etc.) can cause BORSD, OTFCS, or hybrid phenotypes, firmly supporting their status as allelic disorders. Crucially, all reported OTFCS patients with EYA1 variants had renal anomalies, a feature previously considered a hallmark of BORSD. We conclude that BORSD and OTFCS constitute a single EYA1-related diagnostic continuum. This reclassification mandates the development of follow-up protocols that integrate renal, otologic, and skeletal surveillance in EYA1-related disorders, including OTFCS, and refines prognostic and genetic counseling.
1. Introduction
Craniofacial syndromes associated with branchial arch anomalies represent a clinically and genetically heterogeneous group of disorders, often characterized by overlapping features that complicate diagnosis and etiological classification [1,2]. Among these, Otofaciocervical syndrome (OTFCS) is a rare genetic disorder first described by Fara et al. in 1967, with fewer than ten cases reported in the literature [3,4]. It is characterized by peculiar craniofacial traits (e.g., long triangular face, broad forehead, narrow nose and mandible, and high arched palate), ear abnormalities (e.g., low-set, cup-shaped ears with prominent conchae and a hypoplastic tragus and lobe) often associated with hearing loss, and shoulder girdle anomalies (sloping shoulders, low-set clavicles, winged scapulae, and trapezius hypoplasia). Skeletal anomalies other than girdle anomalies and nasolacrimal duct defects are frequently reported, whereas neurodevelopmental delay and short stature are observed only in some patients [5,6]. OTFCS shares significant phenotypic overlap with branchiootorenal spectrum disorders (BORSD) [7,8]. Nonetheless, they have been previously described as clinically distinct entities: phenotypic traits such as facial dysmorphisms and shoulder girdle anomalies were considered specific to OTFCS, whereas BORSD were explicitly characterized by functional and structural renal anomalies (Table 1) [9,10].

Table 1.
Genotypic and phenotypic overlapping within the branchiootorenal and Otofaciocervical syndrome spectrum.
Heterozygous variants in EYA1 (EYA transcriptional coactivator and phosphatase 1) account for approximately 40–75% of individuals clinically diagnosed with BORSD [11,12], but have also been reported in OTFCS patients [13,14,15]. Other genes in the Pax-Six-Eya-Dach network (PSEDN) are likewise implicated in both phenotypes. Heterozygous variants in SIX1 (sine oculis homeobox homolog 1) and SIX5 (sine oculis homeobox homolog 5) have been detected in 3.0–45% and 0–3.1% of individuals with BORSD, respectively [16,17,18]. In addition, biallelic PAX1 (paired box 1) variants underlie OTFCS type 2 with T-cell deficiency (OTFCS2) [19,20,21,22], while loss-of-function (LoF) variants in EYA4 (EYA transcriptional coactivator and phosphatase 4) have more recently been reported in a single affected family [4].
Whether OTFCS and BORSD represent distinct nosological entities or instead form part of a broader phenotypic continuum remains unresolved, as the precise genetic basis of OTFCS is not yet fully clarified. Importantly, some individuals with a BORSD diagnosis present with features typical of OTFCS—musculoskeletal and neurodevelopmental involvement—while some OTFCS patients exhibit renal anomalies, suggesting that the two conditions may, at least in part, represent allelic disorders [11,23,24]. This growing body of evidence supports the hypothesis that these disorders may, at least in part, represent allelic conditions.
In this study, we report a novel patient presenting with OTFCS harboring a de novo microdeletion encompassing EYA1 and perform a comprehensive review of all published cases of EYA1-related disorders. By delineating overlaps and distinctions between OTFCS and BORSD, we aim to refine their allelic relationship, improve diagnostic precision, and inform genetic counseling, while contributing to a deeper understanding of the molecular mechanisms underlying these syndromes.
2. Materials and Methods
2.1. Clinical and Molecular Data
Clinical and audiological information was collected for the index patient, including detailed phenotypic characterization with particular attention to branchial, auricular, renal, and neurodevelopmental features. Audiological assessments included the type and degree of hearing loss. Genomic DNA was extracted from peripheral blood samples.
Chromosome microarray analysis (CMA) was performed using the CytoScan XON array (Thermo Fisher Scientific, Waltham, MA, USA).
Multiplex Ligation-dependent Probe Amplification (MLPA) was performed using the SALSA MLPA probemix P153-B1 EYA1 kit (MRC-Holland, Amsterdam, The Netherlands), and variant analysis was carried out with Coffalyser.Net software v.240129.1959 (MRC-Holland, Amterdam, The Netherlands). The coordinates of detected deletions were mapped to the human genome assembly hg38 (GRCh38). Segregation analysis was performed to determine the inheritance pattern.
2.2. Literature Review and Data Extraction
A systematic literature review was conducted (last search: August 2025) using PubMed, Scopus, Embase, and Google Scholar, with the following keywords: “BORSD”, “BOR syndrome”, “BO syndrome”, “OFC syndrome”, “OTFC syndrome”, “BOF syndrome”, “BOU syndrome”, “branchio-oto-renal”, “branchio-otic”, “Otofaciocervical”, “del”, “deletion”, and “EYA1”. Filters applied: English language, human studies, and original clinical/genetic data.
2.3. Inclusion and Exclusion Criteria
Included: case reports, series, or cohorts with (i) EYA1 SNVs/indels or CNVs and (ii) patient-level clinical data covering ≥2 domains (branchial, otologic, renal, craniofacial, musculoskeletal). Excluded: reviews, functional-only/animal studies, or overlapping cohorts (retaining the most complete report).
2.4. Screening and Data Extraction
Two reviewers independently screened titles/abstracts, followed by a full-text review. Extracted data included demographics, clinical features (categorized as BORSD-typical or OTFCS-typical), variant type (missense, truncating, splice, indel, stop-loss, structural/CNV), and deletion coordinates. All variants were described according to HGVS nomenclature using the EYA1 transcript NM_000503.6 and mapped to GRCh38. Duplicates were removed.
3. Clinical Presentation
A 22-year-old female, born at term, second child of healthy non-consanguineous parents, presented with severe congenital bilateral mixed hearing loss, bilateral preauricular fistulas, hypoplasia of the left shoulder muscles, winged scapula, short stature (<3rd percentile), and a history of speech delay. Chromosome analysis revealed a normal female karyotype (46,XX). Analysis of the EYA1 gene was negative for variants using sequencing approaches. MLPA analysis identified a heterozygous de novo deletion encompassing the entire coding region of EYA1 at 8q13.3. CMA (Figure 1) confirmed a 2.3 Mb interstitial deletion at 8q13.2q13.3 chromosomal region, which spanned from nucleotides 69,068,130 to 71,362,732 (GRCh38) and involved 12 genes (LINC01592, LINC01603, SULF1, SLCO5A1, PRDM14, NCOA2, LOC101926892, TRAM1, LACTB2-AS1, LACTB2, XKR9, EYA1), and which are further characterized in Table 2. The microdeletion occurred de novo because both parents were wild-type.
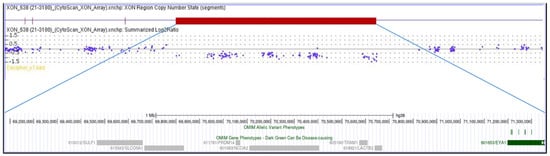
Figure 1.
The molecular karyotype of the novel patient and the associated genes, adapted from the UCSC Genome Browser according to the International System for Human Cytogenetic Nomenclature (ISCN 2024): arr[GRCh38] 8q13.2q13.3(69,068,130_71,362,732)×1.
Table 2.
OMIM genes within the patient’s deleted chromosomal region. pHaplo dosage sensitivity index is presented, where high ranks (i.e., 0.8–1.0) indicate a gene is more likely to exhibit haploinsufficiency and low ranks (i.e., 0.0–0.20) indicate a gene is more likely not to exhibit haploinsufficiency.
4. Results
The search retrieved more than 200 records in PubMed and additional records in Scopus, Embase and Google Scholar; after deduplication and eligibility screening, 55 studies and 141 reported SNVs were included, as described in Supplementary Materials. Among these, 54 (38.3%) were frameshift variants (fs), 30 (21.3%) were nonsense variants (ns), 28 (19.9%) were splice-site variants (sp), 26 (18.4%) were missense variants (ms), 2 (1.4%) were stop-loss/stop-like variants (sl), and 1 (0.7%) was annotated as an indel (Figure 2). EYA1 gene SNVs found in the literature in association with OTFCS/BORSD spectrum are shown in Table 3, according to the first accession of genotype and/or complete phenotype. 1 splice site variant was associated with a single OTFCS case, 1 missense variant was associated with unrelated OTFCS and BORSD cases, while all the remainder SNVs were detected within the BORSD spectrum. Renal involvement is apparently absent in association with 34/141 (24.1%) described SNVs, regardless of the type of variant (ns, fs, sp, ms, sl) within the BORSD spectrum. OTFCS cases (both due to SNVs and CNVs) are further characterized in Table 4 and compared to the reported case.
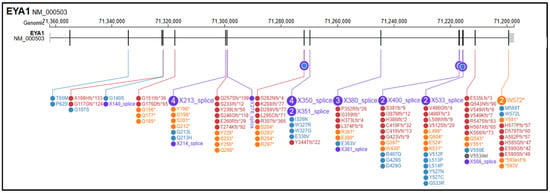
Figure 2.
Graphical representation of variants in the EYA1 (NM_000503.6) gene reported in medical and scientific literature (PubMed, Scopus, Google Scholar) in association with BORSD/OTFCS phenotypes. In yellow, frameshift variants; in purple, splicing variants; in red, nonsense variants; in blue, missense variants; in black, stop-loss variants; in grey, in/del variants. Variant visualization was generated using ProteinPaint Release version: 2.147.0-ecd631c6f (St. Jude Children’s Research Hospital, Memphis, TN, USA; https://proteinpaint.stjude.org (accessed on 25 September 2025).
Table 3.
EYA1 variants have been reported in patients with branchiootorenal spectrum disorders (BORSD), branchiootic (BO) syndrome, or otofaciocervical syndrome (OTFCS). Variants are described according to the HGVS nomenclature, using the reference transcript NM_000503.6 (EYA1) and mapped to the human genome assembly GRCh38. Variant types are classified as missense (ms), nonsense (ns), frameshift (fs), splice (sp), insertion/deletion (indel), or stoploss (sl). Clinical diagnoses are reported as indicated in the original publications, grouped into BOR, BO, OTFCS, or overlapping phenotypes. Only molecularly confirmed cases with sufficient clinical description were included. References correspond to the first report of each genotype–phenotype association.
Table 4.
Reported patients with otofaciocervical syndrome (OTFCS) carrying EYA1 (NM_000503.6) variants, compared with the current case. Clinical features are grouped into core domains: HL = hearing loss; BA = branchial anomalies; EA = external ear anomalies; RA = renal anomalies; MSK = musculoskeletal anomalies; NDD = neurodevelopmental delay; ST = short stature. Additional findings are listed under “Other”. Variants are described according to NM_000503.6 (EYA1) and mapped to the GRCh38 assembly. Variant type was classified as single-nucleotide variant (SNV) or copy-number variant (CNV). Inheritance is indicated when available.
In addition to the distribution of variant classes, our analysis revealed that large EYA1 deletions are enriched among BORSD cases, accounting for approximately 20% of the reports in the literature. Moreover, two-thirds of reported EYA1 SNVs were predicted to be LoF, consistent with haploinsufficiency as the main disease mechanism.
5. Discussion
The present review highlights the complex relationship between BORSD and OTFCS, both associated with EYA1 copy number and sequence variants. BORSD has traditionally been defined by a triad of branchial, otologic, and renal anomalies [7,18]. In contrast, OTFCS has been described as a distinct condition, characterized by musculoskeletal anomalies such as scapular dysplasia and short stature [9,14]. However, our systematic analysis and the present case emphasize that considerable phenotypic overlap exists, and that classical BORSD features may co-occur with OTFCS hallmarks.
The EYA proteins are components of a conserved regulatory network that is often referred to as the PAX–SIX–EYA–DACH developmental network (PSEDN) to better reflect the proteins involved [69]. This network plays a key regulatory role in the early development of multiple organs [70,71]. Notably, all known disease genes implicated in BORSD and OTFCS belong to this network. While OTFCS has also been genetically linked to PAX1 [4,14] and, in a limited number of patients, EYA4 in [19], EYA1 remains the major gene implicated in conditions.
Pathogenic EYA1 variants encompass truncating, missense, splice-site, stop-loss, and copy-number alterations, and have been documented in association with BORSD, OTFCS, and intermediate phenotypes [6,62]. Within our case series, one SNV in the EYA1 gene was described exclusively in association with OTFCS (1/141; 0.7%), one SNV was described in association with both OTFCS and BORSD (1/141; 0.7%), while the remainder (139/141; 98.6%) were described within the BORSD spectrum, with or without renal involvement. Thus, the variant class alone may be insufficient to predict the clinical presentation. This supports the view that haploinsufficiency is the predominant disease mechanism [72,73], but additional genetic or environmental modifiers likely influence phenotypic expressivity. Importantly, the observation that OTFCS can also result from missense and splice variants, and not exclusively from large deletions, further challenges the concept of OTFCS as a purely contiguous gene deletion syndrome [9,14]. Complex rearrangements, inversions, and insertions further contribute to the mutational spectrum [74,75].
A particularly noteworthy finding from our review is that the majority of published patients with OTFCS due to EYA1 defects presented with renal anomalies, while in approximately 25% of EYA1-related cases of BORSD, they were absent regardless of the variant type. Since renal involvement has been traditionally associated with BORSD, this observation undermines the concept of a strict clinical separation between the two syndromes. Instead, it suggests that musculoskeletal involvement in OTFCS and renal anomalies in BORSD are not mutually exclusive, but somewhat variable manifestations of the same allelic defect. Conversely, OTFCS caused by other genetic mechanisms may represent distinct subtypes, supporting the concept of a subclassification of OTFCS according to the underlying molecular etiology.
To our knowledge, this is the first reported case of OTFCS due to an EYA1 defect characterized by Chromosomal Microarray (CMA). This provides a precise genomic definition of the 8q13.2q13.3 microdeletion underlying the phenotype and allows a detailed assessment of co-deleted genes. We also acknowledge that the interpretation of microdeletion cases is complicated by the involvement of multiple genes. In our case, the 8q13.2q13.3 deletion spans additional genes besides EYA1. We therefore reviewed their known or predicted functions. Although some have roles in developmental pathways, none have been consistently implicated in renal or musculoskeletal phenotypes resembling BORSD or OTFCS. Thus, while we cannot completely rule out modifying effects from neighboring genes, the current evidence supports EYA1 haploinsufficiency as the principal driver of both the craniofacial–musculoskeletal anomalies and the renal phenotype in our patient.
The wide spectrum of presentations of EYA1-related disorders suggests that modifying factors, such as genetic background, environmental influences, or stochastic events during development, may critically modulate the expressivity of EYA1 variants [76]. Analogous patterns are well recognized in other genetic conditions such as COL2A1 (Collagen, Type II, Alpha-1)-related skeletal dysplasias and TBX6 (T-Box Transcription Factor 6)-related segmentation defects, where allelic heterogeneity and modifiers account for wide phenotypic variability [77,78,79,80]. Rather than being distinct syndromes, BORSD and OTFCS may represent different clinical expressions of EYA1 dysfunction within the context of the broader PSEDN. Reports of identical or highly similar EYA1 anomalies resulting in divergent phenotypes in different families further support this model [35,72,81].
From a clinical standpoint, acknowledging OTFCS and BORSD as allelic disorders has significant implications. It underscores the need to systematically evaluate musculoskeletal and developmental features in patients diagnosed with BORSD, and conversely, to ensure comprehensive renal and auditory assessments in patients with OTFCS. Grouping both under the umbrella of EYA1-related disorders would enhance and streamline variant interpretation, strengthen genetic counseling, and support the development of follow-up protocols that integrate renal, otologic, and skeletal surveillance.
Future studies should pursue three main directions: (i) large-scale genotype–phenotype analyses integrating both BORSD and OTFCS cases; (ii) functional studies to elucidate the molecular impact of different EYA1 variants; and (iii) investigation of potential second-site modifiers within the PSEDN Network that might influence phenotypic outcome.
6. Conclusions
Our findings consolidate the model of BORSD and OTFCS as allelic disorders within a unified EYA1-related spectrum. This reclassification is critical for clinical practice: it improves diagnostic accuracy, mandates comprehensive phenotyping—most notably, systematic renal screening in all OTFCS patients—and refines prognostic and genetic counseling. Future research integrating deep phenotyping, genomic data, and functional studies will be essential to elucidate the mechanisms underlying the striking phenotypic variability within this spectrum.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes16111267/s1, Supplementary File: Flow diagram for literature review and data extraction via databases and registers.
Author Contributions
Conceptualization, L.G. and M.L.C.; Methodology, L.G.; Software, B.A.; Validation, O.P., S.M. and M.B.; Formal Analysis, O.P. and B.A.; Investigation, M.B.; Resources, G.N.; Data Curation, O.P.; Writing—Original Draft Preparation, L.G. and M.L.C.; Writing—Review & Editing, M.C. and G.N.; Visualization, M.L.C.; Supervision, M.C. and M.B.; Project Administration, G.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable. Ethical approval was waived for this study. No supplementary analysis was performed on the patients, except for the diagnostic genetic test.
Informed Consent Statement
The study was conducted according to the guidelines of the Declaration of Helsinki. We obtained written consent from the patients beforehand, as required by our regulations. The human samples used in this study were acquired from a by-product of routine care or industry.
Data Availability Statement
All datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors would like to thank the family for their generous participation in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Johnson, J.M.; Moonis, G.; Green, G.E.; Carmody, R.; Burbank, H.N. Syndromes of the First and Second Branchial Arches, Part 1: Embryology and Characteristic Defects. Am. J. Neuroradiol. 2011, 32, 14–19. [Google Scholar] [CrossRef]
- Adams, A.; Mankad, K.; Offiah, C.; Childs, L. Branchial Cleft Anomalies: A Pictorial Review of Embryological Development and Spectrum of Imaging Findings. Insights Imaging 2016, 7, 69–76. [Google Scholar] [CrossRef]
- Fára, M.; Chlupácková, V.; Hrivnákova, J. Familial oto-facio-cervical dysmorphia. Acta Chir. Orthop. Traumatol. Cech. 1967, 34, 511–520. [Google Scholar]
- Gana, S.; Valetto, A.; Toschi, B.; Sardelli, I.; Cappelli, S.; Peroni, D.; Bertini, V. Familial Interstitial 6q23.2 Deletion Including Eya4 Associated With Otofaciocervical Syndrome. Front. Genet. 2019, 10, 650. [Google Scholar] [CrossRef]
- Salinas-Torres, V.M.; Salinas-Torres, R.A. Otofaciocervical Syndrome and Metachondromatosis in a Girl: Presentation of a Novel Association and Remarks on Clinical Variability of Branchial-Arch Disorders. Int. J. Pediatr. Otorhinolaryngol. 2016, 85, 19–21. [Google Scholar] [CrossRef]
- Castiglione, A.; Melchionda, S.; Carella, M.; Trevisi, P.; Bovo, R.; Manara, R.; Martini, A. EYA1-Related Disorders: Two Clinical Cases and a Literature Review. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1201–1210. [Google Scholar] [CrossRef]
- Smith, R.J. Branchiootorenal Spectrum Disorder. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Cho, S.H.; Jeong, S.H.; Choi, W.H.; Lee, S.-Y. Genomic Landscape of Branchio-Oto-Renal Syndrome through Whole-Genome Sequencing: A Single Rare Disease Center Experience in South Korea. Int. J. Mol. Sci. 2024, 25, 8149. [Google Scholar] [CrossRef]
- Dallapiccola, B.; Mingarelli, R. Otofaciocervical Syndrome: A Sporadic Patient Supports Splitting from the Branchio-Oto-Renal Syndrome. J. Med. Genet. 1995, 32, 816–818. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vm, S.-T.; Rivera, H. Branchiootorenal Syndrome with Skeletal Defects: A Novel Association in a Mexican Child. Clin. Dysmorphol. 2015, 24, 21–23. [Google Scholar] [CrossRef]
- Krug, P.; Morinière, V.; Marlin, S.; Koubi, V.; Gabriel, H.D.; Colin, E.; Bonneau, D.; Salomon, R.; Antignac, C.; Heidet, L. Mutation Screening of the EYA1, SIX1, and SIX5 Genes in a Large Cohort of Patients Harboring Branchio-Oto-Renal Syndrome Calls into Question the Pathogenic Role of SIX5 Mutations. Hum. Mutat. 2011, 32, 183–190. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, S.; Qiu, Y.; Xing, Q.; Lu, W. A Novel Mutation in EYA1 in a Chinese Family with Branchio-Oto-Renal Syndrome. BMC Med. Genet. 2018, 19, 139. [Google Scholar] [CrossRef] [PubMed]
- Estefanía, E.; Ramírez-Camacho, R.; Gomar, M.; Trinidad, A.; Arellano, B.; García-Berrocal, J.R.; Verdaguer, J.M.; Vilches, C. Point Mutation of an EYA1-Gene Splice Site in a Patient with Oto-Facio-Cervical Syndrome. Ann. Hum. Genet. 2006, 70, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Rickard, S.; Van’T Hoff, W.; Barnicoat, A.; Russell-Eggitt, I.; Winter, R.; Bitner-Glindzicz, M.; Parker, M. Oto-Facio-Cervical (OFC) Syndrome Is a Contiguous Gene Deletion Syndrome Involving EYA1: Molecular Analysis Confirms Allelism with BOR Syndrome and Further Narrows the Duane Syndrome Critical Region to 1 cM. Hum. Genet. 2001, 108, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Mercer, C.; Gilbert, R.; Loughlin, S.; Foulds, N. Patient with an EYA1 Mutation with Features of Branchio-Oto-Renal and Oto-Facio-Cervical Syndrome. Clin. Dysmorphol. 2006, 15, 211–212. [Google Scholar] [CrossRef]
- Kochhar, A.; Orten, D.J.; Sorensen, J.L.; Fischer, S.M.; Cremers, C.W.R.J.; Kimberling, W.J.; Smith, R.J.H. SIX1 Mutation Screening in 247 Branchio-Oto-Renal Syndrome Families: A Recurrent Missense Mutation Associated with BOR. Hum. Mutat. 2008, 29, 565. [Google Scholar] [CrossRef]
- Hoskins, B.E.; Cramer, C.H.; Silvius, D.; Zou, D.; Raymond, R.M.; Orten, D.J.; Kimberling, W.J.; Smith, R.J.H.; Weil, D.; Petit, C.; et al. Transcription Factor SIX5 Is Mutated in Patients with Branchio-Oto-Renal Syndrome. Am. J. Hum. Genet. 2007, 80, 800–804. [Google Scholar] [CrossRef]
- Chang, E.H.; Menezes, M.; Meyer, N.C.; Cucci, R.A.; Vervoort, V.S.; Schwartz, C.E.; Smith, R.J.H. Branchio-Oto-Renal Syndrome: The Mutation Spectrum in EYA1 and Its Phenotypic Consequences. Hum. Mutat. 2004, 23, 582–589. [Google Scholar] [CrossRef]
- Pohl, E.; Aykut, A.; Beleggia, F.; Karaca, E.; Durmaz, B.; Keupp, K.; Arslan, E.; Palamar, M.; Yigit, G.; Özkinay, F.; et al. A Hypofunctional PAX1 Mutation Causes Autosomal Recessively Inherited Otofaciocervical Syndrome. Hum. Genet. 2013, 132, 1311–1320. [Google Scholar] [CrossRef]
- Paganini, I.; Sestini, R.; Capone, G.L.; Putignano, A.L.; Contini, E.; Giotti, I.; Gensini, F.; Marozza, A.; Barilaro, A.; Porfirio, B.; et al. A Novel PAX1 Null Homozygous Mutation in Autosomal Recessive Otofaciocervical Syndrome Associated with Severe Combined Immunodeficiency. Clin. Genet. 2017, 92, 664–668. [Google Scholar] [CrossRef]
- Sherlaw-Sturrock, C.; Austin, T.; Baptista, J.; Gilmour, K.; Naik, S. Dysmorphism and Immunodeficiency-One of the Differential Diagnoses Is PAX1 Related Otofaciocervical Syndrome Type 2. Eur. J. Med. Genet. 2022, 65, 104523. [Google Scholar] [CrossRef]
- Elbagoury, N.M.; Abdel-Aleem, A.F.; Sharaf-Eldin, W.E.; Ashaat, E.A.; Esswai, M.L. A Novel Truncating Mutation in PAX1 Gene Causes Otofaciocervical Syndrome Without Immunodeficiency. J. Mol. Neurosci. MN 2023, 73, 976–982. [Google Scholar] [CrossRef]
- Stockley, T.L.; Mendoza-Londono, R.; Propst, E.J.; Sodhi, S.; Dupuis, L.; Papsin, B.C. A Recurrent EYA1 Mutation Causing Alternative RNA Splicing in Branchio-Oto-Renal Syndrome: Implications for Molecular Diagnostics and Disease Mechanism. Am. J. Med. Genet. 2009, 149A, 322–327. [Google Scholar] [CrossRef]
- Unzaki, A.; Morisada, N.; Nozu, K.; Ye, M.J.; Ito, S.; Matsunaga, T.; Ishikura, K.; Ina, S.; Nagatani, K.; Okamoto, T.; et al. Clinically Diverse Phenotypes and Genotypes of Patients with Branchio-Oto-Renal Syndrome. J. Hum. Genet. 2018, 63, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, W.K.K.; Liang, Q.; Zhang, N.; He, J.; Li, X.; Zhang, X.; Xu, L.; Chan, M.T.V.; Ng, S.S.M.; et al. Disruption of NCOA2 by Recurrent Fusion with LACTB2 in Colorectal Cancer. Oncogene 2016, 35, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Liang, X.; Sun, X.; Chen, H.; Dong, Y.; Wu, L.; Gu, S.; Han, S. Nuclear Receptor Coactivator 2 Promotes Human Breast Cancer Cell Growth by Positively Regulating the MAPK/ERK Pathway. Front. Oncol. 2019, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, N.; Toyota, M.; Suzuki, H.; Honma, T.; Fujikane, T.; Ohmura, T.; Nishidate, T.; Ohe-Toyota, M.; Maruyama, R.; Sonoda, T.; et al. Gene Amplification and Overexpression of PRDM14 in Breast Cancers. Cancer Res. 2007, 67, 9649–9657. [Google Scholar] [CrossRef]
- Isidor, B.; Pichon, O.; Redon, R.; Day-Salvatore, D.; Hamel, A.; Siwicka, K.A.; Bitner-Glindzicz, M.; Heymann, D.; Kjellén, L.; Kraus, C.; et al. Mesomelia-Synostoses Syndrome Results from Deletion of SULF1 and SLCO5A1 Genes at 8q13. Am. J. Hum. Genet. 2010, 87, 95–100. [Google Scholar] [CrossRef]
- Lai, J.; Chien, J.; Staub, J.; Avula, R.; Greene, E.L.; Matthews, T.A.; Smith, D.I.; Kaufmann, S.H.; Roberts, L.R.; Shridhar, V. Loss of HSulf-1 up-Regulates Heparin-Binding Growth Factor Signaling in Cancer. J. Biol. Chem. 2003, 278, 23107–23117. [Google Scholar] [CrossRef]
- Görlich, D.; Hartmann, E.; Prehn, S.; Rapoport, T.A. A Protein of the Endoplasmic Reticulum Involved Early in Polypeptide Translocation. Nature 1992, 357, 47–52. [Google Scholar] [CrossRef]
- Orten, D.J.; Fischer, S.M.; Sorensen, J.L.; Radhakrishna, U.; Cremers, C.W.R.J.; Marres, H.A.M.; Van Camp, G.; Welch, K.O.; Smith, R.J.H.; Kimberling, W.J. Branchio-Oto-Renal Syndrome (BOR): Novel Mutations in the EYA1 Gene, and a Review of the Mutational Genetics of BOR. Hum. Mutat. 2008, 29, 537–544. [Google Scholar] [CrossRef]
- Lee, J.D.; Kim, S.-C.; Koh, Y.W.; Lee, H.-J.; Choi, S.-Y.; Kim, U.-K. A Novel Frameshift Mutation in the EYA1 Gene in a Korean Family with Branchio-Oto-Renal Syndrome. Ann. Clin. Lab. Sci. 2009, 39, 303–306. [Google Scholar]
- Kim, H.R.; Song, M.H.; Kim, M.-A.; Kim, Y.-R.; Lee, K.-Y.; Sonn, J.K.; Lee, J.; Choi, J.Y.; Kim, U.-K. Identification of a Novel Nonsynonymous Mutation of EYA1 Disrupting Splice Site in a Korean Patient with BOR Syndrome. Mol. Biol. Rep. 2014, 41, 4321–4327. [Google Scholar] [CrossRef]
- Wang, S.-H.; Wu, C.-C.; Lu, Y.-C.; Lin, Y.-H.; Su, Y.-N.; Hwu, W.-L.; Yu, I.-S.; Hsu, C.-J. Mutation Screening of the EYA1, SIX1, and SIX5 Genes in an East Asian Cohort with Branchio-Oto-Renal Syndrome. Laryngoscope 2012, 122, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Klingbeil, K.D.; Greenland, C.M.; Arslan, S.; Llamos Paneque, A.; Gurkan, H.; Demir Ulusal, S.; Maroofian, R.; Carrera-Gonzalez, A.; Montufar-Armendariz, S.; Paredes, R.; et al. Novel EYA1 Variants Causing Branchio-Oto-Renal Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2017, 98, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Ideura, M.; Nishio, S.; Moteki, H.; Takumi, Y.; Miyagawa, M.; Sato, T.; Kobayashi, Y.; Ohyama, K.; Oda, K.; Matsui, T.; et al. Comprehensive Analysis of Syndromic Hearing Loss Patients in Japan. Sci. Rep. 2019, 9, 11976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, J.; Wang, H.; Liu, M.; Lu, S.; Xu, H.; Tang, W.; Zheng, G. Novel Likely Pathogenic Variant in the EYA1 Gene Causing Branchio Oto Renal Syndrome and the Exploration of Pathogenic Mechanisms. BMC Med. Genomics 2024, 17, 89. [Google Scholar] [CrossRef]
- Riedhammer, K.M.; Ćomić, J.; Tasic, V.; Putnik, J.; Abazi-Emini, N.; Paripovic, A.; Stajic, N.; Meitinger, T.; Nushi-Stavileci, V.; Berutti, R.; et al. Exome Sequencing in Individuals with Congenital Anomalies of the Kidney and Urinary Tract (CAKUT): A Single-Center Experience. Eur. J. Hum. Genet. 2023, 31, 674–680. [Google Scholar] [CrossRef]
- Tang, P.; Li, J.; Li, J.; Yang, J.; Zhu, J. Prenatal Diagnosis and Genetic Analysis of a Fetus with Branchio-Oto-Renal Syndrome: A Case Report. Medicine 2022, 101, e31172. [Google Scholar] [CrossRef]
- Kumar, S.; Deffenbacher, K.; Cremers, C.W.; Van Camp, G.; Kimberling, W.J. Branchio-Oto-Renal Syndrome: Identification of Novel Mutations, Molecular Characterization, Mutation Distribution, and Prospects for Genetic Testing. Genet. Test. 1997, 1, 243–251. [Google Scholar] [CrossRef]
- Wang, H.H.; Sun, J.W.; Wan, G.L.; Chen, H.; Li, W.J.; Zhao, W.; Pan, C.C. A new pathogenic variation of EYA1 gene in a family with BOR syndrome and the diagnostic exploration. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2020, 55, 1069–1072. [Google Scholar] [CrossRef]
- Rickard, S.; Boxer, M.; Trompeter, R.; Bitner-Glindzicz, M. Importance of Clinical Evaluation and Molecular Testing in the Branchio-Oto-Renal (BOR) Syndrome and Overlapping Phenotypes. J. Med. Genet. 2000, 37, 623–627. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanggaard, K.M.; Rendtorff, N.D.; Kjaer, K.W.; Eiberg, H.; Johnsen, T.; Gimsing, S.; Dyrmose, J.; Nielsen, K.O.; Lage, K.; Tranebjaerg, L. Branchio-Oto-Renal Syndrome: Detection of EYA1 and SIX1 Mutations in Five out of Six Danish Families by Combining Linkage, MLPA and Sequencing Analyses. Eur. J. Hum. Genet. 2007, 15, 1121–1131. [Google Scholar] [CrossRef]
- Abdelhak, S.; Kalatzis, V.; Heilig, R.; Compain, S.; Samson, D.; Vincent, C.; Weil, D.; Cruaud, C.; Sahly, I.; Leibovici, M.; et al. A Human Homologue of the Drosophila Eyes Absent Gene Underlies Branchio-Oto-Renal (BOR) Syndrome and Identifies a Novel Gene Family. Nat. Genet. 1997, 15, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Song, M.H.; Kwon, T.-J.; Kim, H.R.; Jeon, J.H.; Baek, J.-I.; Lee, W.-S.; Kim, U.-K.; Choi, J.Y. Mutational Analysis of EYA1, SIX1 and SIX5 Genes and Strategies for Management of Hearing Loss in Patients with BOR/BO Syndrome. PLoS ONE 2013, 8, e67236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kwon, M.-J.; Boo, S.H.; Kim, H.-J.; Cho, Y.-S.; Chung, W.-H.; Hong, S.H. A Novel Splice Site Mutation in the EYA1 Gene in a Korean Family with Branchio-Oto (BO) Syndrome. Acta Otolaryngol. 2009, 129, 688–693. [Google Scholar] [CrossRef]
- Masuda, M.; Kanno, A.; Nara, K.; Mutai, H.; Morisada, N.; Iijima, K.; Morimoto, N.; Nakano, A.; Sugiuchi, T.; Okamoto, Y.; et al. Phenotype-Genotype Correlation in Patients with Typical and Atypical Branchio-Oto-Renal Syndrome. Sci. Rep. 2022, 12, 969. [Google Scholar] [CrossRef]
- Chen, A.; Ling, J.; Peng, X.; Liu, X.; Mao, S.; Chen, Y.; Qin, M.; Zhang, S.; Bai, Y.; Song, J.; et al. A Novel EYA1 Mutation Causing Alternative RNA Splicing in a Chinese Family with Branchio-Oto Syndrome: Implications for Molecular Diagnosis and Clinical Application. Clin. Exp. Otorhinolaryngol. 2023, 16, 342–358. [Google Scholar] [CrossRef]
- Okada, M.; Fujimaru, R.; Morimoto, N.; Satomura, K.; Kaku, Y.; Tsuzuki, K.; Nozu, K.; Okuyama, T.; Iijima, K. EYA1 and SIX1 Gene Mutations in Japanese Patients with Branchio-Oto-Renal (BOR) Syndrome and Related Conditions. Pediatr. Nephrol. 2006, 21, 475–481. [Google Scholar] [CrossRef]
- Xing, Z.-K.; Wang, S.-Y.; Xia, X.; Ding, W.-J.; Duan, L.; Cui, X.; Xu, B.-C.; Zhu, Y.-M.; Liu, X.-W. Targeted Next-Generation Sequencing Identifies a Novel Frameshift EYA1 Variant Causing Branchio-Otic Syndrome in a Chinese Family. Int. J. Pediatr. Otorhinolaryngol. 2020, 138, 110202. [Google Scholar] [CrossRef]
- Spruijt, L.; Hoefsloot, L.H.; van Schaijk, G.H.W.H.; van Waardenburg, D.; Kremer, B.; Brackel, H.J.L.; de Die-Smulders, C.E.M. Identification of a Novel EYA1 Mutation Presenting in a Newborn with Laryngomalacia, Glossoptosis, Retrognathia, and Pectus Excavatum. Am. J. Med. Genet. 2006, 140, 1343–1345. [Google Scholar] [CrossRef]
- Lin, Z.; Li, J.; Pei, Y.; Mo, Y.; Jiang, X.; Chen, L. Misdiagnosed Branchio-Oto-Renal Syndrome Presenting as Proteinuria and Renal Insufficiency with Insidious Signs since Early Childhood: A Report of Three Cases. BMC Nephrol. 2023, 24, 248. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef]
- Ma, J.; Huang, R.; Ma, X.L.; Li, X.; Zhang, T.S.; Ruan, B. Identification and genetic analysis of new mutations in EYA1 gene of BOS syndrome. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2021, 56, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Yalcouyé, A.; Traoré, O.; Diarra, S.; Schrauwen, I.; Esoh, K.; Kadlubowska, M.K.; Bharadwaj, T.; Adadey, S.M.; Kéita, M.; Guinto, C.O.; et al. A Monoallelic Variant in EYA1 Is Associated with Branchio-Otic Syndrome in a Malian Family. Mol. Genet. Genom. Med. 2022, 10, e1995. [Google Scholar] [CrossRef] [PubMed]
- Namba, A.; Abe, S.; Shinkawa, H.; Kimberling, W.J.; Usami, S.I. Genetic Features of Hearing Loss Associated with Ear Anomalies: PDS and EYA1 Mutation Analysis. J. Hum. Genet. 2001, 46, 518–521. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bałdyga, N.; Oziębło, D.; Gan, N.; Furmanek, M.; Leja, M.L.; Skarżyński, H.; Ołdak, M. The Genetic Background of Hearing Loss in Patients with EVA and Cochlear Malformation. Genes 2023, 14, 335. [Google Scholar] [CrossRef]
- Li, G.; Shen, Q.; Sun, L.; Liu, H.; An, Y.; Xu, H. A de Novo and Novel Mutation in the EYA1 Gene in a Chinese Child with Branchio-Oto-Renal Syndrome. Intractable Rare Dis. Res. 2018, 7, 42–45. [Google Scholar] [CrossRef]
- Nardi, E.; Palermo, A.; Cusimano, P.; Mulè, G.; Cerasola, G. Young Woman with Branchio-Oto-Renal Syndrome and a Novel Mutation in the EYA-1 Gene. Clin. Nephrol. 2011, 76, 330–333. [Google Scholar] [CrossRef]
- Gigante, M.; d’Altilia, M.; Montemurno, E.; Diella, S.; Bruno, F.; Netti, G.S.; Ranieri, E.; Stallone, G.; Infante, B.; Grandaliano, G.; et al. Branchio-Oto-Renal Syndrome (BOR) Associated with Focal Glomerulosclerosis in a Patient with a Novel EYA1 Splice Site Mutation. BMC Nephrol. 2013, 14, 60. [Google Scholar] [CrossRef][Green Version]
- Chen, P.; Liu, H.; Lin, Y.; Xu, J.; Zhu, W.; Wu, H.; Yang, T. EYA1 Mutations Leads to Branchio-Oto Syndrome in Two Chinese Han Deaf Families. Int. J. Pediatr. Otorhinolaryngol. 2019, 123, 141–145. [Google Scholar] [CrossRef]
- Tian, Y.; Lv, Y.; Wang, H.; Che, J.; Cui, F.; Guo, J.; Tian, W.; Peng, J.; Yang, B.; Li, H.; et al. Prenatal Phenotypic Analysis of Branchio-Oto-Renal Spectrum Disorder Attributable to EYA1 Gene Pathogenic Variants and Systematic Literature Review. Prenat. Diagn. 2024, 44, 1509–1517. [Google Scholar] [CrossRef]
- Spahiu, L.; Merovci, B.; Ismaili Jaha, V.; Batalli Këpuska, A.; Jashari, H. Case Report of a Novel Mutation of the EYA1 Gene in a Patient with Branchio-Oto-Renal Syndrome. Balk. J. Med. Genet. 2016, 19, 91–94. [Google Scholar] [CrossRef]
- He, J.; Mahmoudi, A.; Gu, Y.; Fu, J.; Yuan, Q.; Liu, W. Case Report: A Novel Mutation in the EYA1 Gene in a Child with Branchiootic Syndrome with Secretory Otitis Media and Bilateral Vestibular Hypofunction. Front. Genet. 2023, 14, 1292085. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.F.; Xu, G.E.; Chen, B.; Sun, S.P.; Zeng, B.P.; Tang, W.X.; Lu, W. Branchio-oto-renal syndrome or branchio-oto syndrome: The clinical and genetic analysis in five Chinese families. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2022, 57, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Li, Y.; Zhao, H.; Lin, K.; Gao, Y.; Ming, C.; Ma, J. Clinical phenotypic and genetic analysis of syndrome families with EYA1 gene variants. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi = J. Clin. Otorhinolaryngol. Head Neck Surg. 2024, 38, 636–640. [Google Scholar] [CrossRef]
- Matsunaga, T.; Okada, M.; Usami, S.-I.; Okuyama, T. Phenotypic Consequences in a Japanese Family Having Branchio-Oto-Renal Syndrome with a Novel Frameshift Mutation in the Gene EYA1. Acta Otolaryngol. 2007, 127, 98–104. [Google Scholar] [CrossRef]
- Vincent, C.; Kalatzis, V.; Compain, S.; Levilliers, J.; Slim, R.; Graia, F.; Pereira, M.L.; Nivelon, A.; Croquette, M.F.; Lacombe, D. A Proposed New Contiguous Gene Syndrome on 8q Consists of Branchio-Oto-Renal (BOR) Syndrome, Duane Syndrome, a Dominant Form of Hydrocephalus and Trapeze Aplasia; Implications for the Mapping of the BOR Gene. Hum. Mol. Genet. 1994, 3, 1859–1866. [Google Scholar] [CrossRef]
- Ikeda, K.; Watanabe, Y.; Ohto, H.; Kawakami, K. Molecular Interaction and Synergistic Activation of a Promoter by Six, Eya, and Dach Proteins Mediated through CREB Binding Protein. Mol. Cell. Biol. 2002, 22, 6759–6766. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhang, H.; Yan, Y.; Mei, Q.; Wu, K. Retinal Determination Gene Networks: From Biological Functions to Therapeutic Strategies. Biomark. Res. 2023, 11, 18. [Google Scholar] [CrossRef]
- Kozmik, Z.; Holland, N.D.; Kreslova, J.; Oliveri, D.; Schubert, M.; Jonasova, K.; Holland, L.Z.; Pestarino, M.; Benes, V.; Candiani, S. Pax–Six–Eya–Dach Network during Amphioxus Development: Conservation in Vitro but Context Specificity in Vivo. Dev. Biol. 2007, 306, 143–159. [Google Scholar] [CrossRef]
- Au, P.B.; Chernos, J.E.; Thomas, M.A. Review of the Recurrent 8q13.2q13.3 Branchio-oto-renal Related Microdeletion, and Report of an Additional Case with Associated Distal Arthrogryposis. Am. J. Med. Genet. A 2016, 170, 2984–2987. [Google Scholar] [CrossRef]
- Morisada, N.; Nozu, K.; Iijima, K. Branchio-Oto-Renal Syndrome: Comprehensive Review Based on Nationwide Surveillance in Japan. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2014, 56, 309–314. [Google Scholar] [CrossRef]
- Schmidt, T.; Bierhals, T.; Kortüm, F.; Bartels, I.; Liehr, T.; Burfeind, P.; Shoukier, M.; Frank, V.; Bergmann, C.; Kutsche, K. Branchio-Otic Syndrome Caused by a Genomic Rearrangement: Clinical Findings and Molecular Cytogenetic Studies in a Patient with a Pericentric Inversion of Chromosome 8. Cytogenet. Genome Res. 2014, 142, 1–6. [Google Scholar] [CrossRef]
- Masuda, L.; Hasegawa, A.; Kamura, H.; Hasegawa, F.; Yamamura, M.; Taniguchi, K.; Ito, Y.; Hata, K.; Samura, O.; Okamoto, A. Missense BICD2 Variants in Fetuses with Congenital Arthrogryposis and Pterygia. Hum. Genome Var. 2024, 11, 32. [Google Scholar] [CrossRef]
- Sewerin, S.; Aurnhammer, C.; Skubic, C.; Blagotinšek Cokan, K.; Jeruc, J.; Rozman, D.; Pfister, F.; Dittrich, K.; Mayer, B.; Schönauer, R.; et al. Mechanisms of Pathogenicity and the Quest for Genetic Modifiers of Kidney Disease in Branchiootorenal Syndrome. Clin. Kidney J. 2024, 17, sfad260. [Google Scholar] [CrossRef]
- Chen, W.; Lin, J.; Wang, L.; Li, X.; Zhao, S.; Liu, J.; Akdemir, Z.C.; Zhao, Y.; Du, R.; Ye, Y.; et al. TBX6 Missense Variants Expand the Mutational Spectrum in a Non-Mendelian Inheritance Disease. Hum. Mutat. 2020, 41, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Barat-Houari, M.; Dumont, B.; Fabre, A.; Them, F.T.; Alembik, Y.; Alessandri, J.-L.; Amiel, J.; Audebert, S.; Baumann-Morel, C.; Blanchet, P.; et al. The Expanding Spectrum of COL2A1 Gene Variants IN 136 Patients with a Skeletal Dysplasia Phenotype. Eur. J. Hum. Genet. 2016, 24, 992–1000. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Markova, T.; Kenis, V.; Melchenko, E.; Osipova, D.; Nagornova, T.; Orlova, A.; Zakharova, E.; Dadali, E.; Kutsev, S. Clinical and Genetic Characteristics of COL2A1-Associated Skeletal Dysplasias in 60 Russian Patients: Part I. Genes 2022, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Morales-Angulo, C.; del Castillo, I.; Moreno, F.; Moreno-Pelayo, M.A. Stickler and Branchio-Oto-Renal Syndromes in a Patient with Mutations in EYA1 and COL2A1 Genes. Clin. Genet. 2008, 73, 262–267. [Google Scholar] [CrossRef]
- Brophy, P.D.; Alasti, F.; Darbro, B.W.; Clarke, J.; Nishimura, C.; Cobb, B.; Smith, R.J.; Manak, J.R. Genome-Wide Copy Number Variation Analysis of a Branchio-Oto-Renal Syndrome Cohort Identifies a Recombination Hotspot and Implicates New Candidate Genes. Hum. Genet. 2013, 132, 1339–1350. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).