
Identification and Functional Analysis of Candidate Genes Influencing Citrus Leaf Size Through Transcriptome and Coexpression Network Approaches

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction, cDNA Library Preparation, and Sequencing
2.3. Identification of Differentially Expressed Genes
2.4. Construction of the Coexpression Network
2.5. qRT-PCR
3. Results
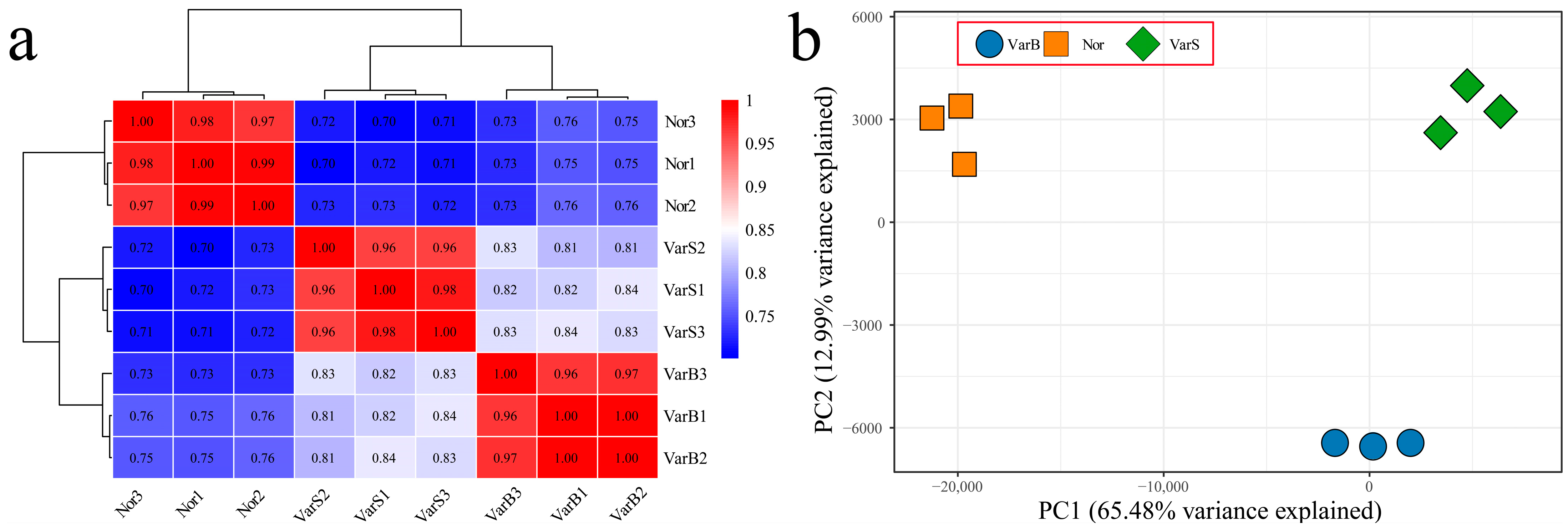
3.1. Overall Analysis of RNA-Seq Data
3.2. Differential Expression Analysis
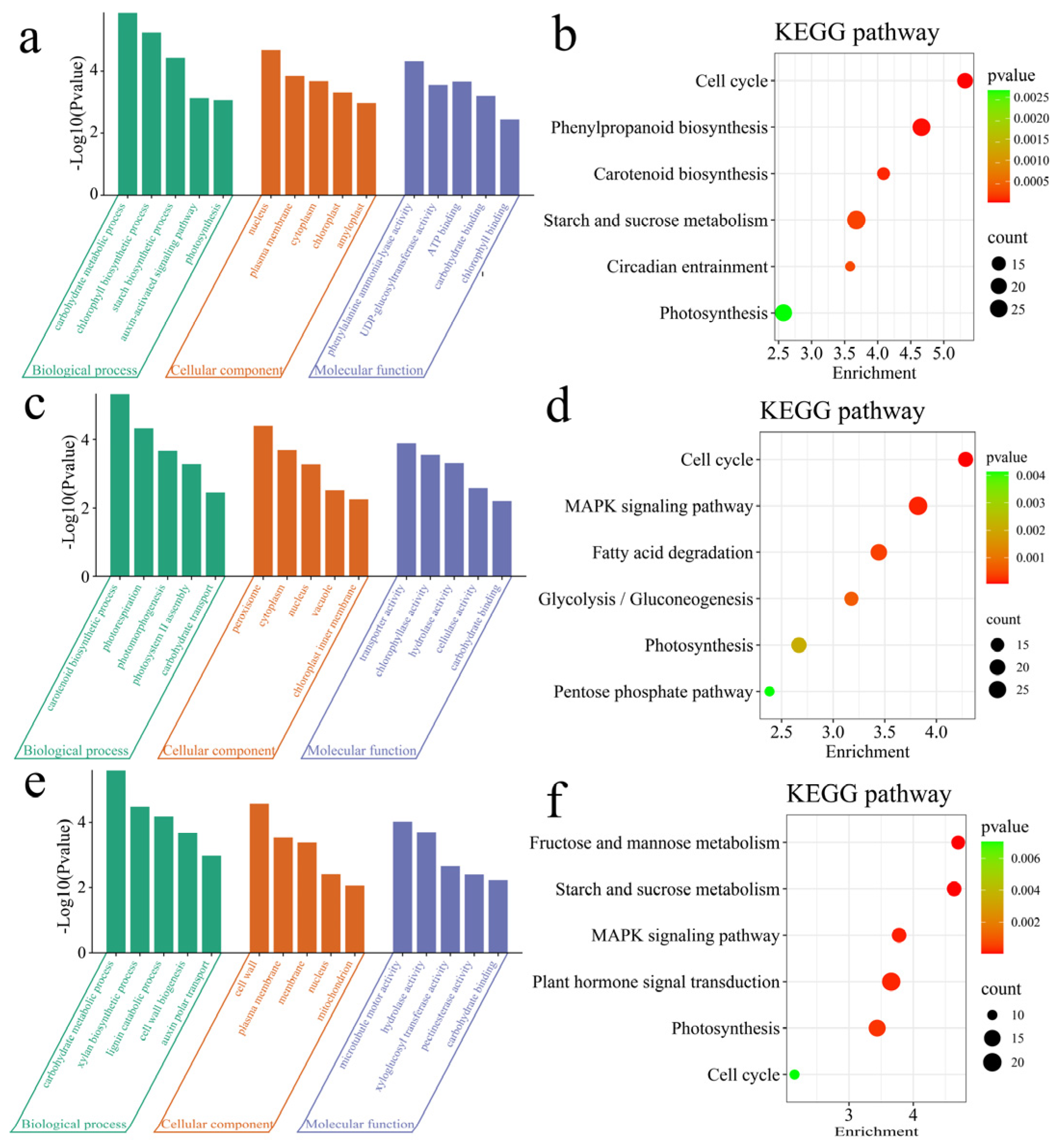
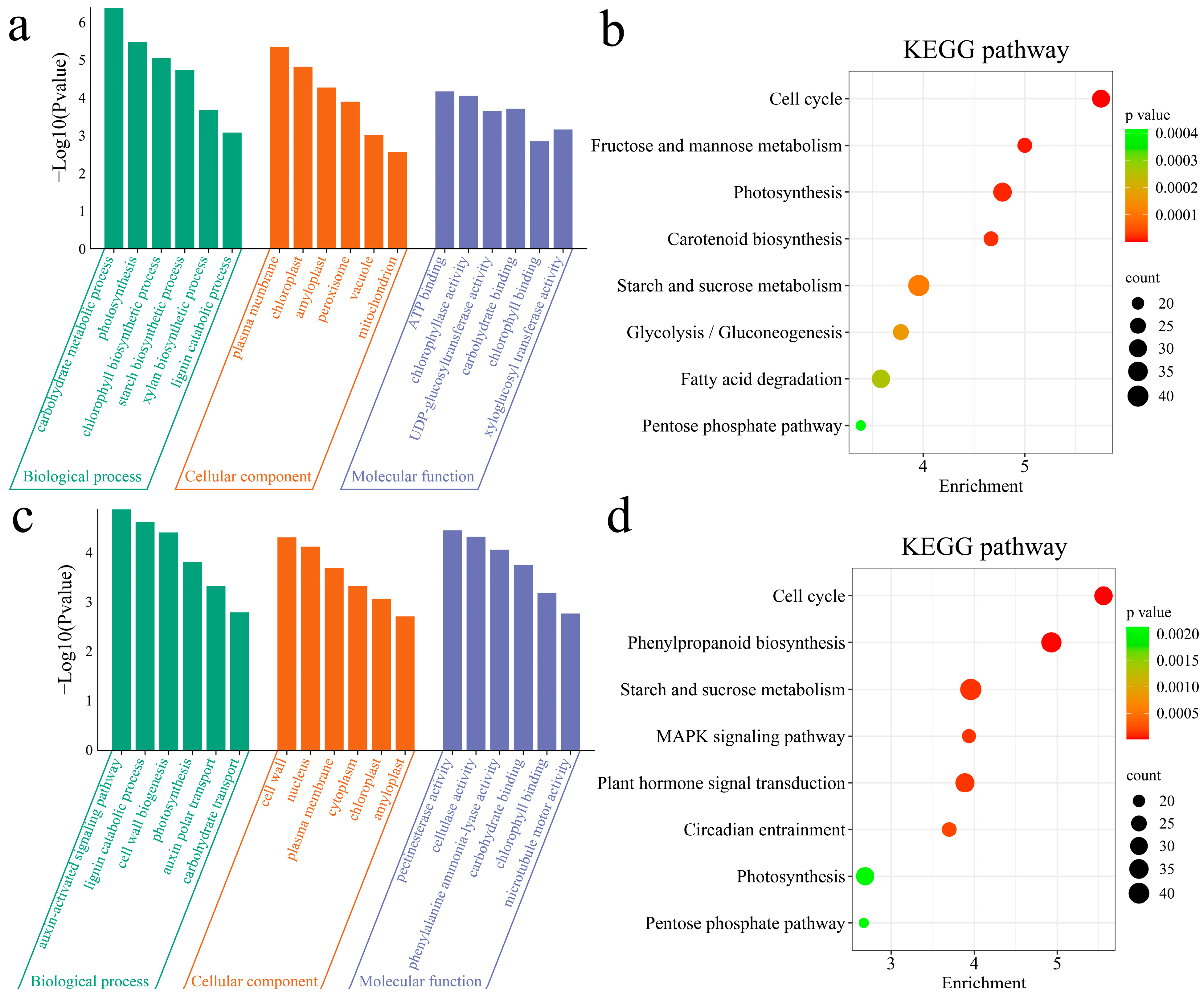
3.3. Enrichment Analysis
3.4. Transcription Factor Analysis
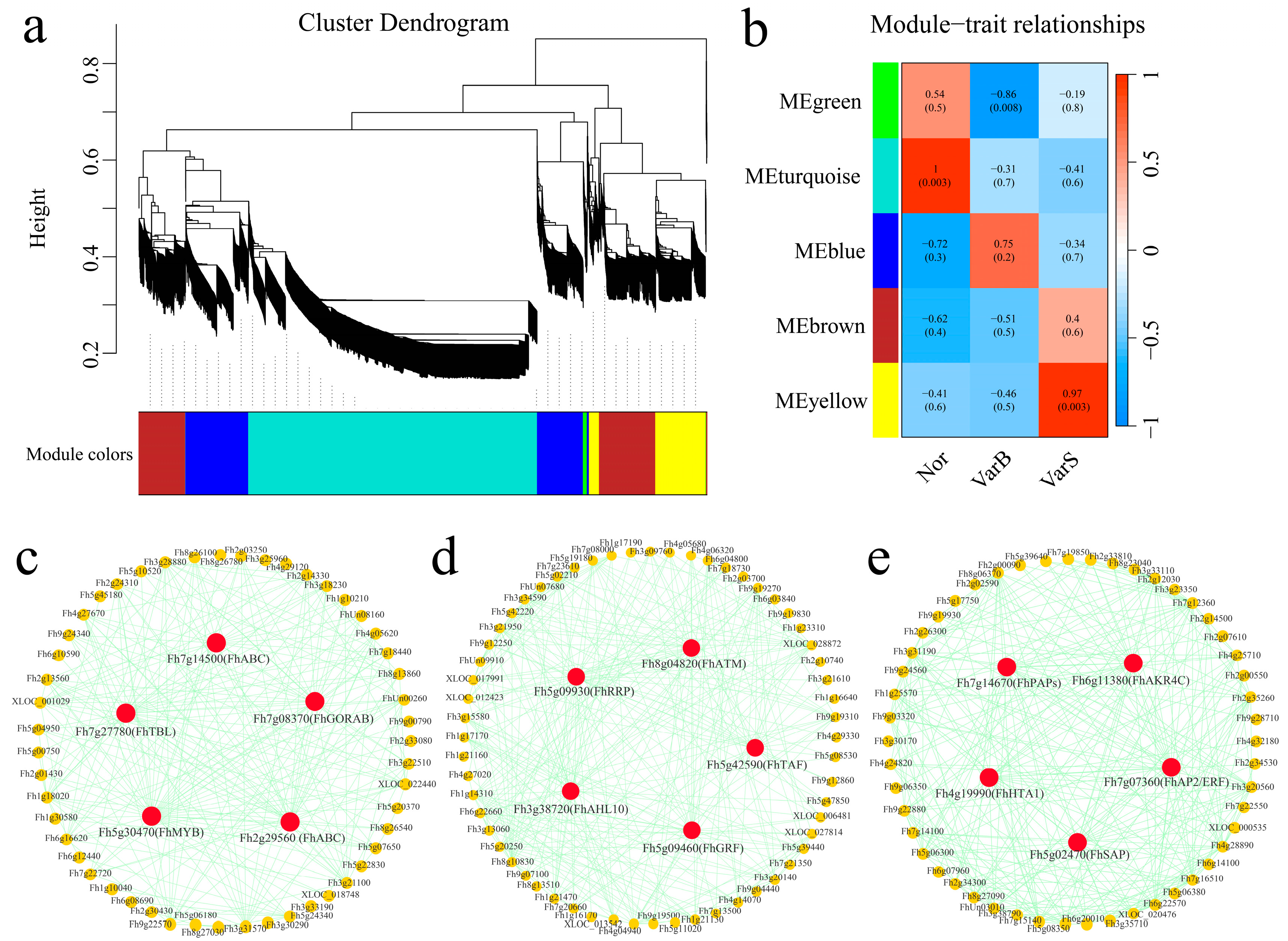
3.5. Construction of a Weighted Gene Coexpression Network
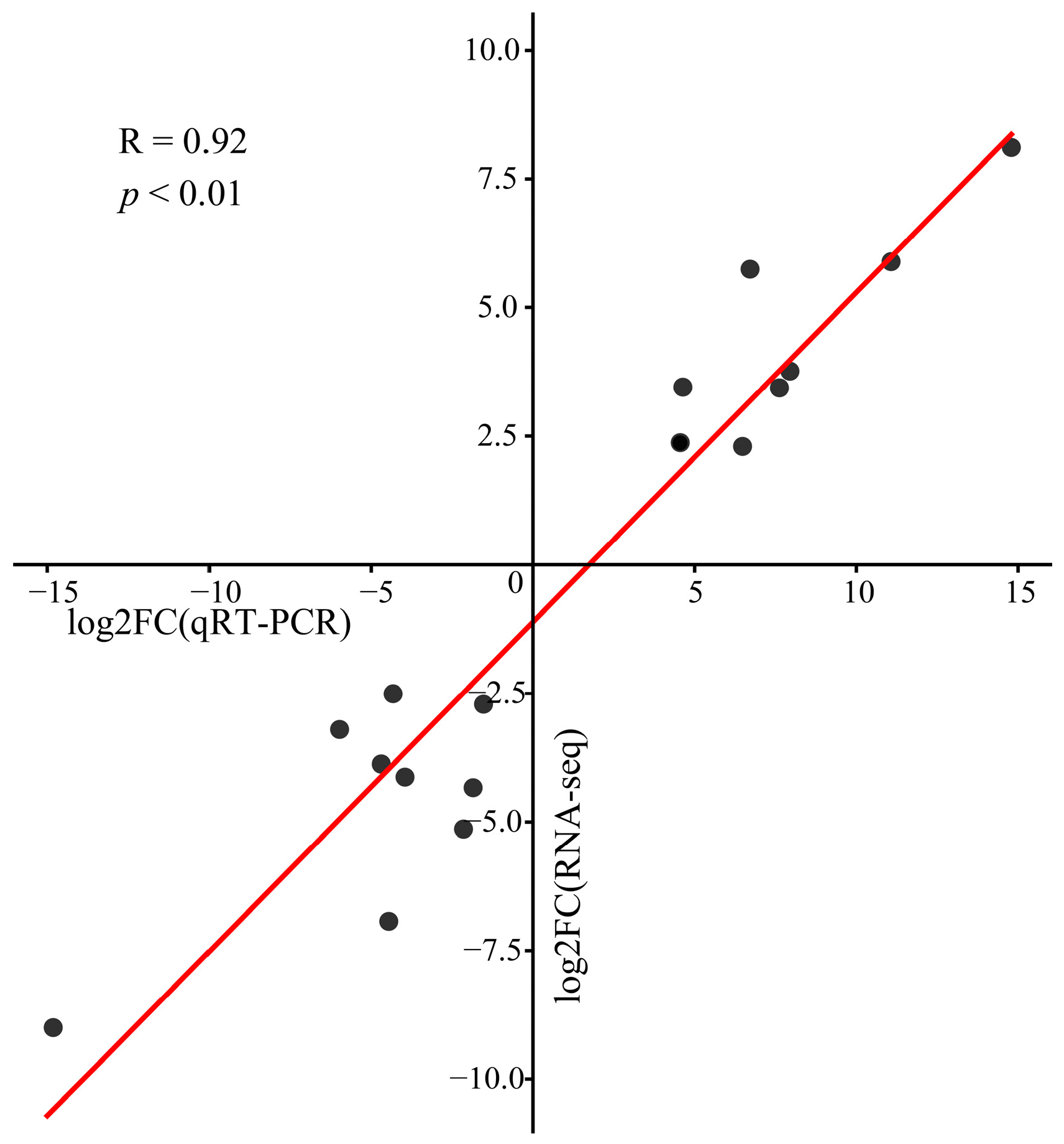
3.6. qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.; Xu, Y.; Li, Y.; Zheng, S.; Zhao, Z.; Chen, M.; Yang, H.; Yi, H.; Wu, J. Transcription factor CsMYB77 negatively regulates fruit ripening and fruit size in citrus. Plant Physiol. 2024, 194, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yuan, G.; Wang, Y.; An, G.; Li, W.; Liu, J.; Sun, D. Mapping and functional verification of leaf yellowing genes in watermelon during whole growth period. Front. Plant Sci. 2022, 13, 1049114. [Google Scholar] [CrossRef]
- Czesnick, H.; Lenhard, M. Size control in plants--Lessons from leaves and flowers. Cold Spring Harb. Perspect. Biol. 2015, 7, a019190. [Google Scholar] [CrossRef] [PubMed]
- Ustin, S.L.; Jacquemoud, S. How the optical properties of leaves modify the absorption and scattering of energy and enhance leaf functionality. Remote Sens. Plant Biodivers. 2020, 349–384. [Google Scholar]
- Umesh, M.R.; Angadi, S.; Begna, S.; Gowda, P.; Prasad, P.V. Shade tolerance response of legumes in terms of biomass accumulation, leaf photosynthesis, and chlorophyll pigment under reduced sunlight. Crop. Sci. 2023, 63, 278–292. [Google Scholar] [CrossRef]
- Bar, M.; Ori, N. Compound leaf development in model plant species. Curr. Opin. Plant Biol. 2015, 23, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, J.; Lenhard, M. Regulation of plant lateral-organ growth by modulating cell number and size. Curr. Opin. Plant Biol. 2014, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bar, M.; Ori, N. Leaf development and morphogenesis. Development 2014, 141, 4219–4230. [Google Scholar] [CrossRef] [PubMed]
- Vercruysse, J.; Baekelandt, A.; Gonzalez, N.; Inzé, D. Molecular networks regulating cell division during Arabidopsis leaf growth. J. Exp. Bot. 2020, 71, 2365–2378. [Google Scholar] [CrossRef]
- Liu, H.; Hu, D.; Du, P.; Wang, L.; Liang, X.; Li, H.; Lu, Q.; Li, S.; Liu, H.; Chen, X.; et al. Single-cell RNA-seq describes the transcriptome landscape and identifies critical transcription factors in the leaf blade of the allotetraploid peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2021, 19, 2261–2276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liang, Z.; Feng, D.; Jiang, S.; Wang, Y.; Du, Z.; Li, R.; Hu, G.; Zhang, P.; Ma, Y.; et al. Transcriptional landscape of rice roots at the single-cell resolution. Mol. Plant. 2021, 14, 384–394. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Feng, L.Y.; Wang, J.H.; Fu, Y.F.; Cai, X.; Wang, C.Q.; Du, J.B.; Yuan, M.; Chen, Y.E.; Xu, P.Z.; et al. Two-factor ANOVA of SSH and RNA-seq analysis reveal development-associated Pi-starvation genes in oilseed rape. Planta 2019, 250, 1073–1088. [Google Scholar] [CrossRef]
- Du, X.; Liu, N.; Lu, P.; Wang, Y.; Lu, B.; Tian, S.; Zhang, Z. RNA-seq-based transcriptome profiling of early fruit development in Chieh-qua and analysis of related transcription factors. Sci. Rep. 2024, 14, 13489. [Google Scholar] [CrossRef] [PubMed]
- Pomares-Viciana, T.; Del Río-Celestino, M.; Román, B.; Die, J.; Pico, B.; Gómez, P. First RNA-seq approach to study fruit set and parthenocarpy in zucchini (Cucurbita pepo L.). BMC Plant Biol. 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.F.; Wu, C.J.; Guo, Y.F.; Walther, D.; Shan, W.; Chen, J.Y.; Chen, L.; Lu, W.J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef]
- Feng, G.; Wu, J.; Xu, Y.; Lu, L.; Yi, H. High-spatiotemporal-resolution transcriptomes provide insights into fruit development and ripening in Citrus sinensis. Plant Biotechnol. J. 2021, 19, 1337–1353. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, J.; Xu, Y.; Zheng, W.; Wang, S.; Chen, P.; Zeng, B.; Yang, S.; Jiang, X.; Liu, Z.; et al. Pangenome analysis provides insight into the evolution of the orange subfamily and a key gene for citric acid accumulation in citrus fruits. Nat. Genet. 2023, 55, 1964–1975. [Google Scholar] [CrossRef]
- Wan, H.; Qiu, H.; Li, Z.; Zhang, X.; Zhang, J.; Jiang, D.; Fernie, A.R.; Lyu, Y.; Cheng, Y.; Wen, W. Transcription factor CsESE3 positively modulates both jasmonic acid and wax biosynthesis in citrus. aBIOTECH 2022, 3, 250–266. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Liao, L.; Deng, L.; Jin, Z.; Huang, Z.; Sun, G.; Xiong, B.; Wang, Z. Combined transcriptome and metabolome analyses reveal candidate genes involved in Tangor (Citrus reticulata × Citrus sinensis) fruit development and quality formation. Int. J. Mol. Sci. 2022, 23, 5457. [Google Scholar] [CrossRef] [PubMed]
- Zandalinas, S.I.; Balfagón, D.; Arbona, V.; Gómez-Cadenas, A. Modulation of antioxidant defense system is associated with combined drought and heat stress tolerance in Citrus. Front. Plant Sci. 2017, 8, 953. [Google Scholar] [CrossRef]
- Zhou, H.; Zeng, R.F.; Liu, T.J.; Ai, X.Y.; Ren, M.K.; Zhou, J.J.; Hu, C.G.; Zhang, J.Z. Drought and low temperature-induced NF-YA1 activates FT expression to promote citrus flowering. Plant Cell Environ. 2022, 45, 3505–3522. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Anido, C.B.; Vatén, A.; Smoot, N.K.; Sharma, N.; Guo, V.; Gong, Y.; Anleu Gil, M.X.; Weimer, A.K.; Bergmann, D.C. Single-cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Dev. Cell 2021, 56, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Skalák, J.; Vercruyssen, L.; Claeys, H.; Hradilová, J.; Černý, M.; Novák, O.; Plačková, L.; Saiz-Fernández, I.; Skaláková, P.; Coppens, F.; et al. Multifaceted activity of cytokinin in leaf development shapes its size and structure in Arabidopsis. Plant J. 2019, 97, 805–824. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Kim, Y.; Shim, Y.; Cho, S.H.; Yang, T.J.; Song, Y.H.; Kang, K.; Paek, N.C. Rice OsGATA16 is a positive regulator for chlorophyll biosynthesis and chloroplast development. Plant J. 2024, 117, 599–615. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yuan, F.; Long, G.; Qin, L.; Deng, Z. Selection of reference genes for quantitative real-time RT-PCR analysis in citrus. Mol. Biol. Rep. 2012, 39, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Fang, Y.; Hu, P.; Tan, Y.; Wang, Y.; Hou, L.; Deng, X.; Wu, H.; Zhu, L.; Zhu, L.; et al. Construction of a high-density genetic map based on SLAF markers and QTL analysis of leaf size in rice. Front. Plant Sci. 2020, 11, 1143. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Bachem, C.W.B.; Helariutta, Y.; Neuhaus, H.E.; Prat, S.; Ruan, Y.L.; Stitt, M.; Sweetlove, L.J.; Tegeder, M.; Wahl, V.; et al. Synchronization of developmental, molecular and metabolic aspects of source-sink interactions. Nat. Plants 2020, 6, 55–66. [Google Scholar] [CrossRef]
- Panda, D.; Biswal, M.; Mohanty, S.; Dey, P.; Swain, A.; Behera, D.; Baig, M.J.; Kumar, A.; Sah, R.P.; Tripathy, B.C.; et al. Contribution of phytochrome a in the regulation of sink capacity starch biosynthesis, grain quality, grain yield and related traits in rice. Plant Arch. 2020, 20, 1179–1194. [Google Scholar]
- Liu, Z.; Nan, Z.; Lin, S.; Meng, W.; Xie, L.; Yu, H.; Zhang, Z.; Wan, S. Organ removal of maize increases peanut canopy photosynthetic capacity, dry matter accumulation, and yield in maize/peanut intercropping. Front. Plant Sci. 2023, 14, 1266969. [Google Scholar] [CrossRef]
- Mehla, S.; Kumar, U.; Kapoor, P.; Singh, Y.; Sihag, P.; Sagwal, V.; Balyan, P.; Kumar, A.; Ahalawat, N.; Lakra, N.; et al. Structural and functional insights into the candidate genes associated with different developmental stages of flag leaf in bread wheat (Triticum aestivum L.). Front. Genet. 2022, 13, 933560. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lu, Z.; Meng, F.; Li, X.; Cong, R.; Ren, T.; Sharkey, T.D.; Lu, J. The reduction in leaf area precedes that in photosynthesis under potassium deficiency: The importance of leaf anatomy. New Phytol. 2020, 227, 1749–1763. [Google Scholar] [CrossRef]
- Chen, J.; Liu, L.; Chen, G.; Wang, S.; Liu, Y.; Zhang, Z.; Li, H.; Wang, L.; Zhou, Z.; Zhao, J.; et al. CsRAXs negatively regulate leaf size and fruiting ability through auxin glycosylation in cucumber. J. Integr. Plant Biol. 2024, 66, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhao, Q.; Chen, L.; Yao, X.; Zhang, W.; Zhang, B.; Xie, F. Effect of drought stress on sugar metabolism in leaves and roots of soybean seedlings. Plant Physiol. Biochem. 2020, 146, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Structure and growth of plant cell walls. Nat. Rev. Mol. Cell Biol. 2024, 25, 340–358. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Pallavi, A.; Gandasi, N.R. Exploring the potential of pheophorbide A, a chlorophyll-derived compound in modulating GLUT for maintaining glucose homeostasis. Front. Endocrinol. 2024, 15, 1330058. [Google Scholar] [CrossRef]
- Zirngibl, M.E.; Araguirang, G.E.; Kitashova, A.; Jahnke, K.; Rolka, T.; Kühn, C.; Nägele, T.; Richter, A.S. Triose phosphate export from chloroplasts and cellular sugar content regulate anthocyanin biosynthesis during high light acclimation. Plant Commun. 2023, 4, 100423. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Shen, J.; Niu, Y.; Zhao, H.; Guo, Y.; Sun, P.; Yang, T.; Jiang, Y.; Zhao, B.; Wang, Z.; et al. Cucumber nuclear factor-YC2/-YC9 target translocon component CsTIC21 in chloroplast photomorphogenesis. Plant Physiol. 2023, 192, 2822–2837. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Jenke, H.; Strauss, S.; Bazakos, C.; Mosca, G.; Lymbouridou, R.; Kierzkowski, D.; Neumann, U.; Naik, P.; Huijser, P.; et al. Cell-cycle-linked growth reprogramming encodes developmental time into leaf morphogenesis. Curr. Biol. 2024, 34, 541–556. [Google Scholar] [CrossRef]
- Vieira, P.; Kantor, M.R.; Jansen, A.; Handoo, Z.A.; Eisenback, J.D. Cellular insights of beech leaf disease reveal abnormal ectopic cell division of symptomatic interveinal leaf areas. PLoS ONE 2023, 18, e0292588. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, L. From G1 to M: A comparative study of methods for identifying cell cycle phases. Brief. Bioinform. 2024, 25, bbad517. [Google Scholar] [CrossRef]
- Soni, N.; Bacete, L. The interplay between cell wall integrity and cell cycle progression in plants. Plant Mol. Biol. 2023, 113, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Zhao, W.; Kong, S.; Li, L.; Lin, S. Overview of molecular mechanisms of plant leaf development: A systematic review. Front. Plant Sci. 2023, 14, 1293424. [Google Scholar] [CrossRef]
- Durand, B.; Gao, F.B.; Raff, M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997, 16, 306–317. [Google Scholar] [CrossRef]
- Shen, W.; Sun, J.; Xiao, Z.; Feng, P.; Zhang, T.; He, G.; Sang, X. Narrow and Stripe Leaf 2 Regulates Leaf Width by Modulating Cell Cycle Progression in Rice. Rice 2023, 16, 20. [Google Scholar] [CrossRef]
- Xiong, R.; Chu, Z.; Peng, X.; Cui, G.; Li, W.; Dong, L. Transcript-wide identification and expression pattern analysis to comprehend the roles of AP2/ERF genes under development and abiotic stress in Trichosanthes kirilowii. BMC Plant Biol. 2023, 23, 354. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, P.; Tang, B.; Hu, Z.; Xie, Q.; Zhou, S.; Chen, G. The AP2/ERF transcription factor SlERF.F5 functions in leaf senescence in tomato. Plant Cell Rep. 2022, 41, 1181–1195. [Google Scholar] [CrossRef]
- Zong, Y.; Hao, Z.; Tu, Z.; Shen, Y.; Zhang, C.; Wen, S.; Yang, L.; Ma, J.; Li, H. Genome-wide survey and identification of AP2/ERF genes involved in shoot and leaf development in Liriodendron chinense. BMC Genom. 2021, 22, 807. [Google Scholar] [CrossRef]
- Marsch-Martinez, N.; Greco, R.; Becker, J.D.; Dixit, S.; Bergervoet, J.H.; Karaba, A.; de Folter, S.; Pereira, A. BOLITA, an Arabidopsis AP2/ERF-like transcription factor that affects cell expansion and proliferation/differentiation pathways. Plant Mol. Biol. 2006, 62, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chai, Z.; Lin, P.; Huang, C.; Huang, G.; Xu, L.; Deng, Z.; Zhang, M.; Zhang, Y.; Zhao, X. Genome-wide identification and expression analysis of AP2/ERF transcription factors in sugarcane (Saccharum spontaneum L.). BMC Genom. 2020, 21, 685. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene id | Arabidopsis thaliana Homologous Gene | Gene Name | Gene Annotation | Source | Leaf Phenotype |
|---|---|---|---|---|---|
| Fh7g14500 | AT3G48770 | ATP-binding cassette transporter proteins (ABC) | Regulates growth and development | VarB | larger leaves |
| Fh7g27780 | AT1G60790 | Trichome birefringence-like (TBL) | Synthesis and deposition of secondary wall cellulose | VarB | larger leaves |
| Fh7g08370 | AT3G52900 | RAB6-interacting golgin (GORAB) | Microtubules required for immunity | VarB | larger leaves |
| Fh5g30470 | AT1G01520 | Transcription Factor (MYB) | Regulates circadian rhythms | VarB | larger leaves |
| Fh2g29560 | AT1G30410 | ATP-binding cassette (ABC) | Regulates growth and development | VarB | larger leaves |
| Fh5g09930 | AT1G12650 | rRNA biogenesis RRP-like protein (RRP) | Processing and capping of rRNA in the nucleus and cytoplasm | Nor | Normal leaves |
| Fh8g04820 | AT2G15570 | Arabidopsis thioredoxin M-type (ATM) | Involved in activating the cell cycle and DNA damage repair | Nor | Normal leaves |
| Fh3g38720 | AT2G33620 | AT-Hook-Like10 (AHL10) | Can inhibit the maturation of shoot apical meristems | Nor | Normal leaves |
| Fh5g42590 | AT1G02680 | TATA-box binding protein associated factor (TAF) | Plays a role in seed development | Nor | Normal leaves |
| Fh5g09460 | AT2G36400 | Growth-regulating factor (GRF) | Plays a role in leaf development | Nor | Normal leaves |
| Fh7g14670 | AT5G23690 | Purple acid phosphatase (PAPs) | Regulates cell wall synthesis | VarS | smaller leaves |
| Fh6g11380 | AT2G37770 | Aldo keto reductase superfamily (AKR4C) | Participates in photosynthesis | VarS | smaller leaves |
| Fh4g19990 | AT5G54640 | HtrA 1 (HTA1) | Response to various adverse stimuli | VarS | smaller leaves |
| Fh7g07360 | AT1G12980 | Transcription Factor (AP2/ERF) | Involved in regulating embryogenesis | VarS | smaller leaves |
| Fh5g02470 | AT5G35770 | Stress-Associated Protein (SAP) | Regulates inflorescence, flower and ovule development | VarS | smaller leaves |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Xiao, Y.; Liu, P.; Pang, Q.; Deng, C.; Fu, C.; Fang, H.; Chen, C. Identification and Functional Analysis of Candidate Genes Influencing Citrus Leaf Size Through Transcriptome and Coexpression Network Approaches. Genes 2025, 16, 97. https://doi.org/10.3390/genes16010097
Wu X, Xiao Y, Liu P, Pang Q, Deng C, Fu C, Fang H, Chen C. Identification and Functional Analysis of Candidate Genes Influencing Citrus Leaf Size Through Transcriptome and Coexpression Network Approaches. Genes. 2025; 16(1):97. https://doi.org/10.3390/genes16010097
Chicago/Turabian StyleWu, Xiaoxiao, Yuanhui Xiao, Ping Liu, Qiuling Pang, Chongling Deng, Cuina Fu, Haimeng Fang, and Chuanwu Chen. 2025. "Identification and Functional Analysis of Candidate Genes Influencing Citrus Leaf Size Through Transcriptome and Coexpression Network Approaches" Genes 16, no. 1: 97. https://doi.org/10.3390/genes16010097
APA StyleWu, X., Xiao, Y., Liu, P., Pang, Q., Deng, C., Fu, C., Fang, H., & Chen, C. (2025). Identification and Functional Analysis of Candidate Genes Influencing Citrus Leaf Size Through Transcriptome and Coexpression Network Approaches. Genes, 16(1), 97. https://doi.org/10.3390/genes16010097