
Genome-Wide Identification and Analysis of the MYC Gene Family in Cotton: Evolution and Expression Profiles During Normal Growth and Stress Response

 ,
,
Abstract
1. Introduction
2. Results
2.1. Genome-Wide Identification of MYC Gene Family in Upland Cotton
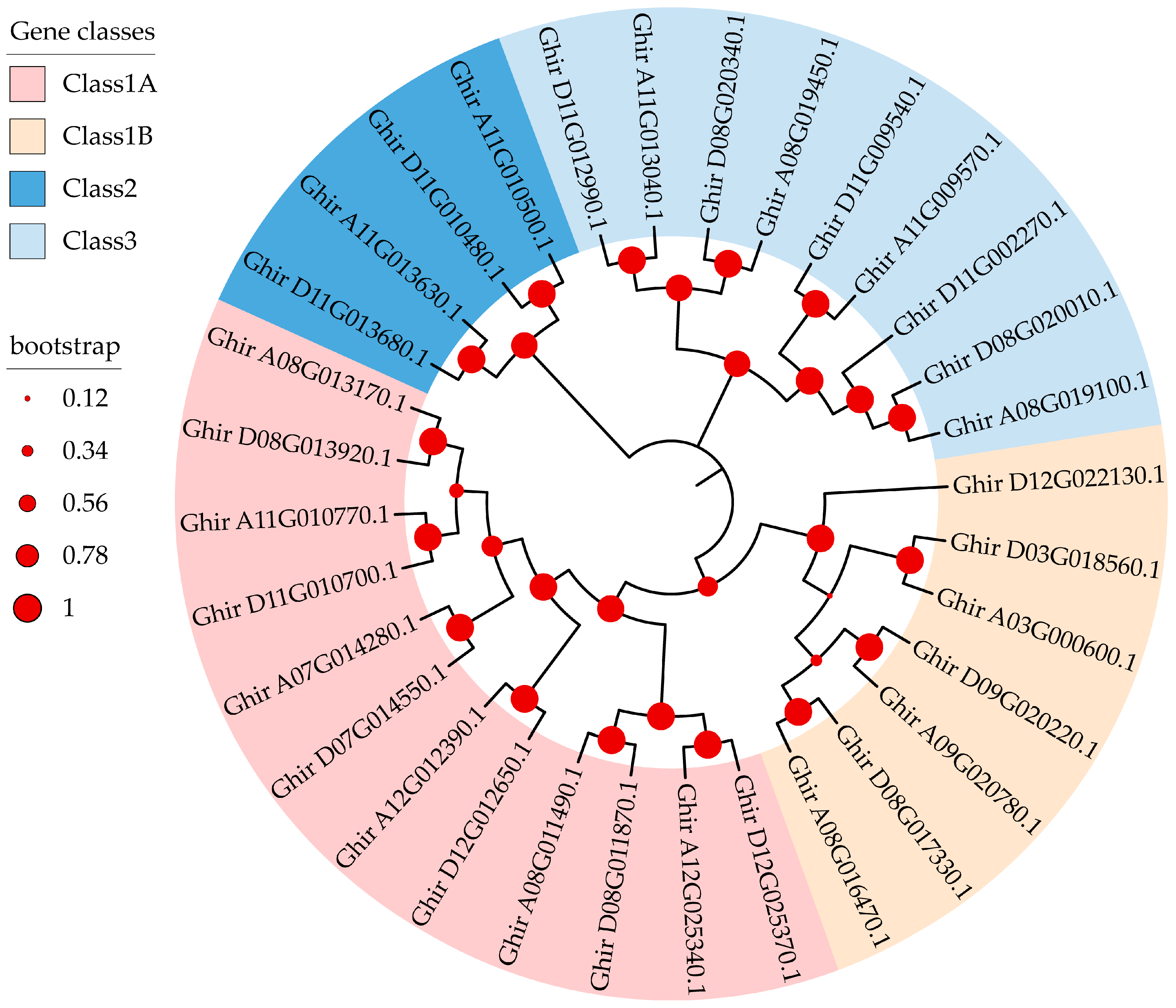
2.2. Phylogenetic Analysis of the MYC Gene Family
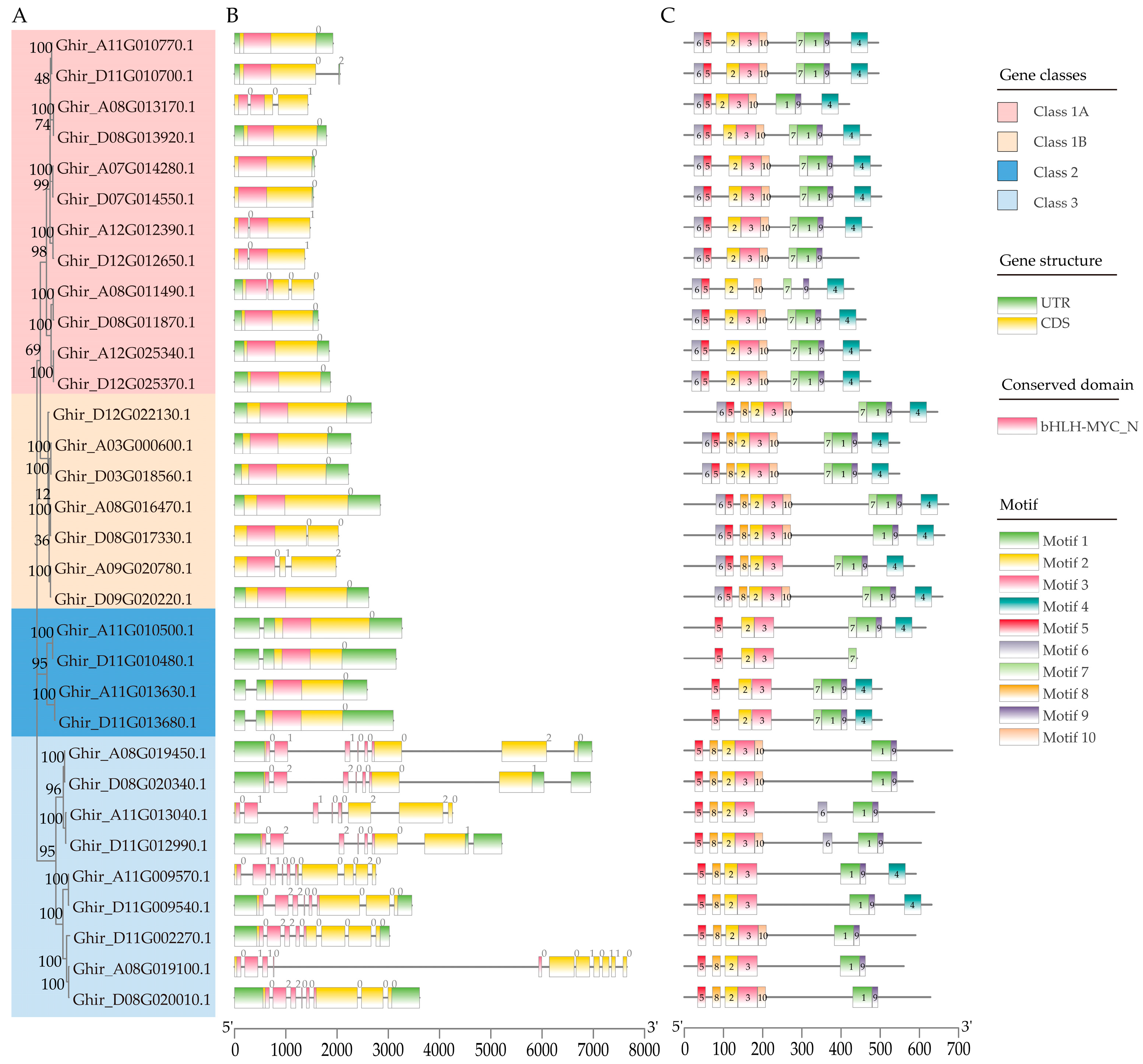
2.3. Analysis of the Structural and Conserved Motifs of GhMYCs
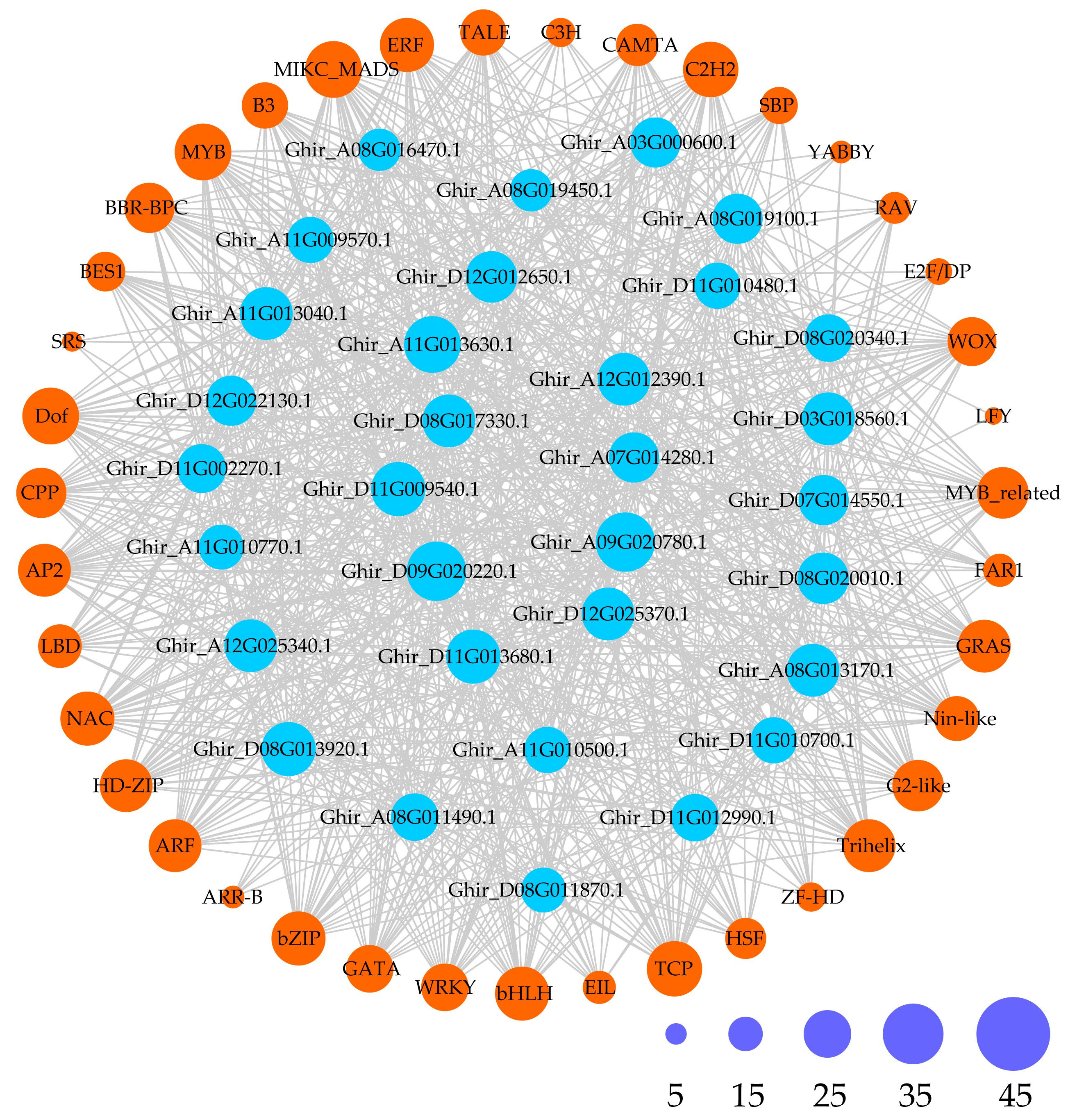
2.4. Prediction of Cis-Acting Elements and Transcription Factors Among the GhMYCs
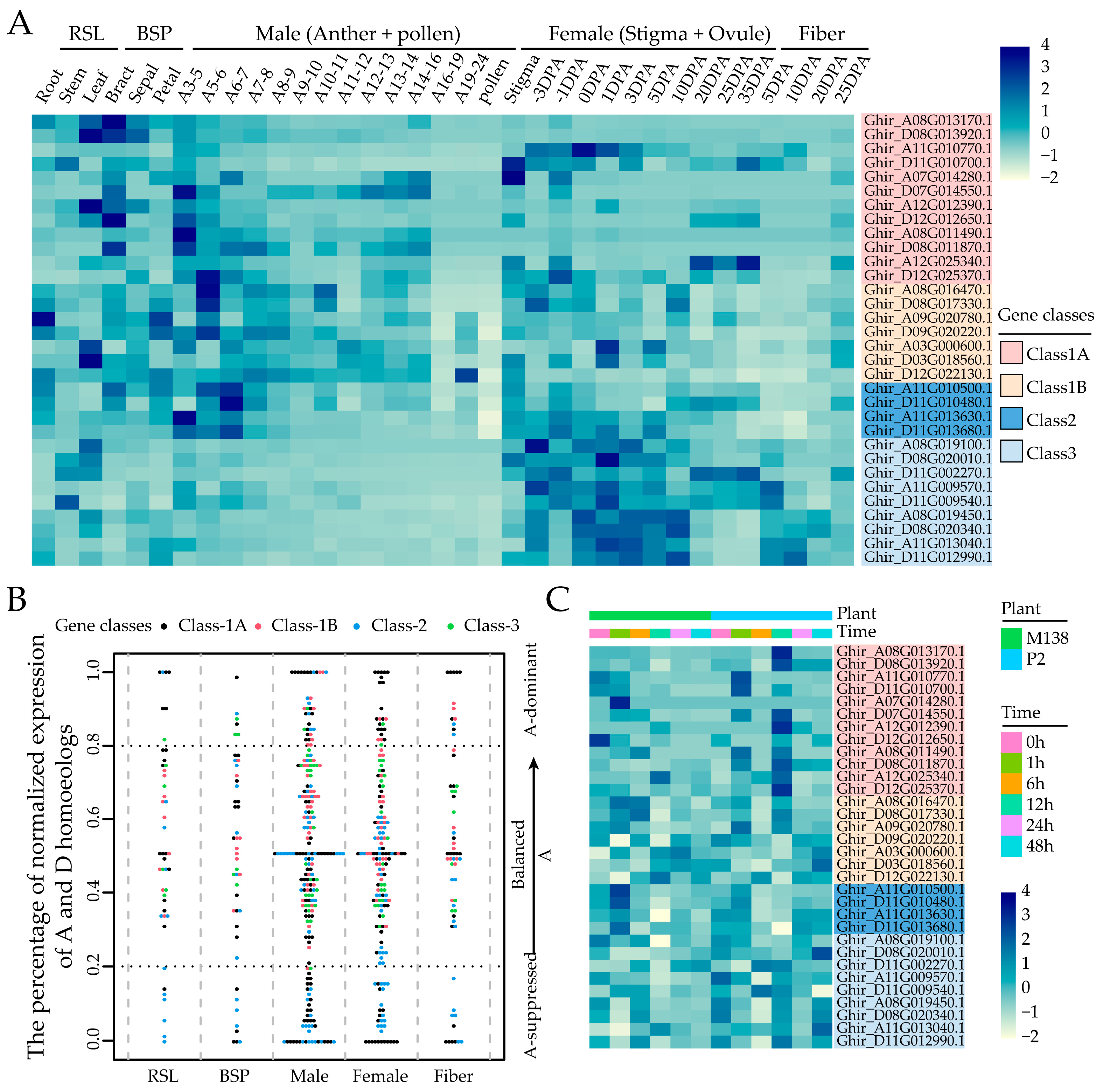
2.5. Tissue Expression Patterns of GhMYCs
2.6. Expression of GhMYCs Under Biotic Stress
3. Discussion
3.1. The Conserved Motif and Sequence Characterization of GhMYCs
3.2. Function Analysis of GhMYC Genes Based on Gene Expression Pattern
4. Conclusions
5. Materials and Methods
5.1. Identification of the MYC Gene Family
5.2. Phylogenetic Analysis and Molecular Evolution Analyses
5.3. Multiple Sequence Alignment of MYC Proteins
5.4. Analysis of Conserved Motifs, Gene Structure, Functional Domains, and 3D Structure
5.5. Promoter Cis-Acting Elements and TF Prediction
5.6. Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lüscher, B.; Eisenman, R.N. New light on Myc and Myb. Part II. Myb. Genes. Dev. 1990, 4, 2235–2241. [Google Scholar] [CrossRef]
- Boter, M.; Ruiz-Rivero, O.; Abdeen, A.; Prat, S. Conserved MYC transcription factors play a key role in jasmonate signaling both in tomato and Arabidopsis. Genes Dev. 2004, 18, 1577–1591. [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Chico, J.M.; Fernández-Calvo, P.; Solano, R. The ZIM domain mediates homo- and heteromeric interactions between Arabidopsis JAZ proteins. Plant J. 2009, 59, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic Acid Signaling Pathway in Plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef]
- Ren, Y.; Zou, W.; Feng, J.; Zhang, C.; Su, W.; Zhao, Z.; Wang, D.; Sun, T.; Wang, W.; Cen, G.; et al. Characterization of the sugarcane MYC gene family and the negative regulatory role of ShMYC4 in response to pathogen stress. Ind. Crops Prod. 2022, 176, 114292. [Google Scholar] [CrossRef]
- Anderson, J.P.; Badruzsaufari, E.; Schenk, P.M.; Manners, J.M.; Desmond, O.J.; Ehlert, C.; Maclean, D.J.; Ebert, P.R.; Kazan, K. Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell 2004, 16, 3460–3479. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F.; Fernandez-Calvo, P.; Zander, M.; Diez-Diaz, M.; Fonseca, S.; Glauser, G.; Lewsey, M.G.; Ecker, J.R.; Solano, R.; Reymond, P. Arabidopsis basic helix-loop-helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell 2013, 25, 3117–3132. [Google Scholar] [CrossRef]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003, 15, 63–78. [Google Scholar] [CrossRef]
- Knight, H.; Zarka, D.G.; Okamoto, H.; Thomashow, M.F.; Knight, M.R. Abscisic acid induces CBF gene transcription and subsequent induction of cold-regulated genes via the CRT promoter element. Plant Physiol. 2004, 135, 1710–1717. [Google Scholar] [CrossRef]
- Verma, D.; Jalmi, S.K.; Bhagat, P.K.; Verma, N.; Sinha, A.K. A bHLH transcription factor, MYC2, imparts salt intolerance by regulating proline biosynthesis in Arabidopsis. FEBS Lett. 2020, 287, 2560–2576. [Google Scholar] [CrossRef]
- Bhadouriya, S.L.; Suresh, A.; Gupta, H.; Mehrotra, S.; Gupta, D.; Mehrotra, R. In Silico Analysis of CCGAC and CATGTG Cis-regulatory Elements Across Genomes Reveals their Roles in Gene Regulation under Stress. Curr. Genom. 2021, 22, 353–362. [Google Scholar] [CrossRef]
- Lahiri, A.; Zhou, L.; He, P.; Datta, A. Detecting drought regulators using stochastic inference in Bayesian networks. PLoS ONE 2021, 16, e0255486. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Y.; Shen, Z.; Wu, M.; Huang, M.; Hong, S.B.; Xu, L.; Zang, Y. Advances in functional studies of plant MYC transcription factors. Theor. Appl. Genet. 2024, 137, 195. [Google Scholar] [CrossRef] [PubMed]
- Uji, Y.; Taniguchi, S.; Tamaoki, D.; Shishido, H.; Akimitsu, K.; Gomi, K. Overexpression of OsMYC2 Results in the Up-Regulation of Early JA-Rresponsive Genes and Bacterial Blight Resistance in Rice. Plant Cell Physiol. 2016, 57, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, M.; Deng, L.; Shen, J.; Fang, M.; Chen, Q.; Lu, Y.; Wang, Q.; Li, C.; Zhai, Q. MYC2 Regulates the Termination of Jasmonate Signaling via an Autoregulatory Negative Feedback Loop. Plant Cell 2019, 31, 106–127. [Google Scholar] [CrossRef]
- Wang, G.; Wang, F.; Xu, Z.; Wang, Y.; Zhang, C.; Zhou, Y.; Hui, F.; Yang, X.; Nie, X.; Zhang, X.; et al. Precise fine-turning of GhTFL1 by base editing tools defines ideal cotton plant architecture. Genome Biol. 2024, 25, 59. [Google Scholar] [CrossRef]
- Johnson, L.Y.D.; Major, I.T.; Chen, Y.; Yang, C.; Vanegas-Cano, L.J.; Howe, G.A. Diversification of JAZ-MYC signaling function in immune metabolism. New Phytol. 2023, 239, 2277–2291. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Li, Y.; Xu, Y.; Wang, Y.; Zhang, R.; Sun, W.; Chen, Q.; Wang, X.J.; Li, C.; et al. MED25 connects enhancer-promoter looping and MYC2-dependent activation of jasmonate signalling. Nat. Plants 2019, 5, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.E.; Acevedo-Acevedo, O.; Miranda, G.S.; Vergara-Barros, P.; Holuigue, L.; Figueroa, C.R.; Figueroa, P.M. Salt stress response triggers activation of the jasmonate signaling pathway leading to inhibition of cell elongation in Arabidopsis primary root. J. Exp. Bot. 2016, 67, 4209–4220. [Google Scholar] [CrossRef]
- Zhao, W.; Huang, H.; Wang, J.; Wang, X.; Xu, B.; Yao, X.; Sun, L.; Yang, R.; Wang, J.; Sun, A.; et al. Jasmonic acid enhances osmotic stress responses by MYC2-mediated inhibition of protein phosphatase 2C1 and response regulators 26 transcription factor in tomato. Plant J. 2023, 113, 546–561. [Google Scholar] [CrossRef]
- Liu, H.; Cui, P.; Zhang, B.; Zhu, J.; Liu, C.; Li, Q. Binding of the transcription factor MYC2-like to the ABRE of the OsCYP2 promoter enhances salt tolerance in Oryza sativa. PLoS ONE 2022, 17, e0276075. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, J.; Xu, J.; Zhang, X.; Xie, Z.; Li, Z. Effect of cold stress on photosynthetic physiological characteristics and molecular mechanism analysis in cold-resistant cotton (ZM36) seedlings. Front. Plant Sci. 2024, 15, 1396666. [Google Scholar] [CrossRef]
- Wang, R.; Yu, M.; Xia, J.; Xing, J.; Fan, X.; Xu, Q.; Cang, J.; Zhang, D. Overexpression of TaMYC2 confers freeze tolerance by ICE-CBF-COR module in Arabidopsis thaliana. Front. Plant Sci. 2022, 13, 1042889. [Google Scholar] [CrossRef]
- Zhao, M.L.; Wang, J.N.; Shan, W.; Fan, J.G.; Kuang, J.F.; Wu, K.Q.; Li, X.P.; Chen, W.X.; He, F.Y.; Chen, J.Y.; et al. Induction of jasmonate signalling regulators MaMYC2s and their physical interactions with MaICE1 in methyl jasmonate-induced chilling tolerance in banana fruit. Plant Cell Environ. 2013, 36, 30–51. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Liu, W.; Wang, N.; Qu, C.; Jiang, S.; Fang, H.; Zhang, Z.; Chen, X. Methyl jasmonate enhances apple’ cold tolerance through the JAZ–MYC2 pathway. Plant Cell Tissue Organ Cult. 2018, 136, 75–84. [Google Scholar] [CrossRef]
- Gao, C.; Qi, S.; Liu, K.; Li, D.; Jin, C.; Li, Z.; Huang, G.; Hai, J.; Zhang, M.; Chen, M. MYC2, MYC3, and MYC4 function redundantly in seed storage protein accumulation in Arabidopsis. Plant Physiol. Biochem. 2016, 108, 63–70. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Guo, Z. Natural uORF variation in plants. Trends Plant Sci. 2024, 29, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, C.; Pan, Z.; Lin, H.; Li, Z.; Hou, X.; Liu, J.; Nie, X.; Wu, Y. Genome-Wide Identification and Analysis of the WNK Kinase Gene Family in Upland Cotton. Plants 2023, 12, 4036. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef]
- Ma, C.; Li, R.; Sun, Y.; Zhang, M.; Li, S.; Xu, Y.; Song, J.; Li, J.; Qi, J.; Wang, L.; et al. ZmMYC2s play important roles in maize responses to simulated herbivory and jasmonate. J. Integr. Plant Biol. 2023, 65, 1041–1058. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Li, Y.; Ma, H.; Chi, H.; Ma, Y.; Yang, J.; Xie, S.; Zhang, R.; Liu, L.; et al. Degradation of de-esterified pctin/homogalacturonan by the polygalacturonase GhNSP is necessary for pollen exine formation and male fertility in cotton. Plant Biotechnol. J. 2022, 20, 1054–1068. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, B.; Sun, Z.; Liu, Z.; Cui, Y.; Ke, H.; Wang, Z.; Wu, L.; Zhang, G.; Wang, G.; et al. A large-scale genomic association analysis identifies a fragment in Dt11 chromosome conferring cotton Verticillium wilt resistance. Plant Biotechn. J. 2021, 19, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Pan, Z.; Aini, N.; Han, P.; Wu, Y.; You, C.; Nie, X. Identification of candidate genes for aphid resistance in upland cotton by QTL mapping and expression analysis. Crop J. 2023, 11, 1600–1604. [Google Scholar] [CrossRef]
- You, J.; Liu, Z.; Qi, Z.; Ma, Y.; Sun, M.; Su, L.; Niu, H.; Peng, Y.; Luo, X.; Zhu, M.; et al. Regulatory controls of duplicated gene expression during fiber development in allotetraploid cotton. Nat. Genet. 2023, 55, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X. version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2006, 23, 127–128. [Google Scholar] [CrossRef]
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Chi, H.; Wei, H.; Wang, H.; Yu, S. Genomewide Identification and Characterization of the Genes Involved in the Flowering of Cotton. Int. J. Mol. Sci. 2022, 23, 7940. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2014, 43, D222–D226. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Niu, R.; Zhou, Y.; Zhang, Y.; Mou, R.; Tang, Z.; Wang, Z.; Zhou, G.; Guo, S.; Yuan, M.; Xu, G. uORFlight: A vehicle toward uORF-mediated translational regulation mechanisms in eukaryotes. Database 2020, 2020, baaa007. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, L.; Li, Y.; Ma, H.; Li, Y.; Ma, Y.; Lv, R.; Yang, J.; Wang, W.; Alifu, A.; et al. Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton. Int. J. Mol. Sci. 2022, 23, 3378. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed]
- Ainin, W.; Yuan-long, W.; Zhen-yuan, P.; Yi-zan, M.; Qiu-shuang, A.; Guang-ling, S.; Pan-xia, S.; Ding-yi, Y.; Hai-rong, L.; Bing-hui, T.; et al. Cotton ethylene response factor GhERF91 involved in the defense against Verticillium dahliae. J. Integr. Agric. 2023, 23, 3328–3342. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Gene Location | Strand | CDS | Protein Length (aa) | Amino Acids MW (kDa) | Theoretical pI | Predicted Localization | Instability Index | Aliphatic Index | Grand Average of Hydropathicity |
|---|---|---|---|---|---|---|---|---|---|---|
| Ghir_A03G000600.1 | Ghir_A03:980039-982310 | + | 1650 | 549 | 60.99 | 5.82 | Nucleus. | 48.55 | 76.36 | −0.60 |
| Ghir_A07G014280.1 | Ghir_A07:29260134-29261695 | - | 1509 | 502 | 56.09 | 5.21 | Nucleus. | 55.4 | 78.03 | −0.45 |
| Ghir_A08G011490.1 | Ghir_A08:78260647-78262193 | + | 1299 | 432 | 48.21 | 6.15 | Chloroplast. Nucleus. | 44.73 | 81.2 | −0.35 |
| Ghir_A08G013170.1 | Ghir_A08:93976363-93977793 | + | 1266 | 421 | 47.05 | 6.22 | Nucleus. | 43.14 | 84.77 | −0.40 |
| Ghir_A08G016470.1 | Ghir_A08:107604743-107607585 | + | 2025 | 674 | 73.57 | 5.46 | Nucleus. | 54.11 | 70.49 | −0.58 |
| Ghir_A08G019100.1 | Ghir_A08:113779847-113787499 | + | 1683 | 560 | 62.40 | 5.78 | Nucleus. | 52.97 | 88.96 | −0.41 |
| Ghir_A08G019450.1 | Ghir_A08:114199928-114206901 | + | 2055 | 684 | 76.48 | 5.05 | Nucleus. | 66.57 | 71.9 | −0.66 |
| Ghir_A09G020780.1 | Ghir_A09:77127717-77129696 | - | 1764 | 587 | 63.77 | 5.52 | Nucleus. | 50.77 | 73.8 | −0.56 |
| Ghir_A11G009570.1 | Ghir_A11:8503876-8506632 | + | 1776 | 591 | 66.83 | 5.37 | Nucleus. | 49.02 | 89.75 | −0.38 |
| Ghir_A11G010500.1 | Ghir_A11:9603951-9607216 | - | 1851 | 616 | 68.18 | 6.19 | Nucleus. | 47.89 | 74.09 | −0.54 |
| Ghir_A11G010770.1 | Ghir_A11:10151540-10153460 | + | 1488 | 495 | 55.19 | 5.37 | Nucleus. | 55.23 | 86.67 | −0.42 |
| Ghir_A11G013040.1 | Ghir_A11:13511016-13515266 | + | 1917 | 638 | 71.78 | 5.83 | Nucleus. | 60.7 | 80.13 | −0.54 |
| Ghir_A11G013630.1 | Ghir_A11:14785560-14788144 | + | 1515 | 504 | 55.69 | 5.51 | Nucleus. | 45.38 | 76.59 | −0.47 |
| Ghir_A12G012390.1 | Ghir_A12:80898036-80899508 | + | 1440 | 479 | 53.46 | 5.19 | Nucleus. | 43.44 | 82.23 | −0.51 |
| Ghir_A12G025340.1 | Ghir_A12:104279639-104281482 | + | 1428 | 475 | 53.05 | 6.35 | Nucleus. | 43.55 | 78.13 | −0.48 |
| Ghir_D03G018560.1 | Ghir_D03:52297122-52299344 | - | 1650 | 549 | 60.70 | 5.96 | Nucleus. | 50.25 | 76.74 | −0.60 |
| Ghir_D07G014550.1 | Ghir_D07:22069145-22070681 | - | 1512 | 503 | 56.22 | 5.2 | Nucleus. | 54.13 | 78.67 | −0.47 |
| Ghir_D08G011870.1 | Ghir_D08:40245698-40247330 | - | 1392 | 463 | 52.00 | 6.24 | Nucleus. | 49.07 | 81.23 | −0.36 |
| Ghir_D08G013920.1 | Ghir_D08:47888044-47889836 | - | 1431 | 476 | 53.07 | 6.36 | Nucleus. | 42.21 | 79.5 | −0.50 |
| Ghir_D08G017330.1 | Ghir_D08:55836510-55838535 | + | 1995 | 664 | 72.32 | 5.29 | Nucleus. | 51.17 | 71.7 | −0.54 |
| Ghir_D08G020010.1 | Ghir_D08:60585556-60589165 | + | 1887 | 628 | 70.24 | 5.95 | Nucleus. | 53.7 | 86.61 | −0.47 |
| Ghir_D08G020340.1 | Ghir_D08:61033485-61040431 | + | 1752 | 583 | 65.29 | 4.97 | Nucleus. | 70.55 | 67.79 | −0.72 |
| Ghir_D09G020220.1 | Ghir_D09:48417384-48420001 | - | 1980 | 659 | 71.78 | 5.21 | Nucleus. | 49.37 | 72.23 | −0.60 |
| Ghir_D11G002270.1 | Ghir_D11:1856954-1859976 | + | 1773 | 590 | 65.88 | 5.73 | Nucleus. | 57.77 | 81.59 | −0.52 |
| Ghir_D11G009540.1 | Ghir_D11:7909491-7912950 | + | 1896 | 631 | 71.21 | 5.48 | Nucleus. | 49.39 | 88.24 | −0.42 |
| Ghir_D11G010480.1 | Ghir_D11:8889334-8892486 | - | 1326 | 441 | 48.81 | 6.81 | Nucleus. | 44.54 | 69.21 | −0.59 |
| Ghir_D11G010700.1 | Ghir_D11:9307960-9310014 | + | 1491 | 496 | 55.45 | 5.35 | Nucleus. | 52.29 | 83.53 | −0.45 |
| Ghir_D11G012990.1 | Ghir_D11:12048161-12053376 | + | 1815 | 604 | 68.51 | 5.25 | Nucleus. | 68.22 | 78.21 | −0.63 |
| Ghir_D11G013680.1 | Ghir_D11:12872213-12875310 | + | 1515 | 504 | 55.57 | 5.59 | Nucleus. | 44.6 | 78.73 | −0.44 |
| Ghir_D12G012650.1 | Ghir_D12:41821932-41823302 | + | 1338 | 445 | 49.55 | 5.32 | Nucleus. | 39.66 | 80.18 | −0.47 |
| Ghir_D12G022130.1 | Ghir_D12:56236545-56239215 | + | 1941 | 646 | 71.00 | 5.47 | Nucleus. | 50.61 | 71.56 | −0.58 |
| Ghir_D12G025370.1 | Ghir_D12:59331943-59333814 | + | 1428 | 475 | 53.04 | 6.4 | Nucleus. | 44.57 | 78.13 | −0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Wang, L.; Wang, X.; Lu, L.; Han, P.; Zhang, C.; Han, M.; Xiang, S.; Wang, H.; Xuan, L.; et al. Genome-Wide Identification and Analysis of the MYC Gene Family in Cotton: Evolution and Expression Profiles During Normal Growth and Stress Response. Genes 2025, 16, 20. https://doi.org/10.3390/genes16010020
Chen J, Wang L, Wang X, Lu L, Han P, Zhang C, Han M, Xiang S, Wang H, Xuan L, et al. Genome-Wide Identification and Analysis of the MYC Gene Family in Cotton: Evolution and Expression Profiles During Normal Growth and Stress Response. Genes. 2025; 16(1):20. https://doi.org/10.3390/genes16010020
Chicago/Turabian StyleChen, Jingxi, Long Wang, Xiufang Wang, Lu Lu, Peng Han, Caidie Zhang, Min Han, Siyu Xiang, Haibiao Wang, Lizhong Xuan, and et al. 2025. "Genome-Wide Identification and Analysis of the MYC Gene Family in Cotton: Evolution and Expression Profiles During Normal Growth and Stress Response" Genes 16, no. 1: 20. https://doi.org/10.3390/genes16010020
APA StyleChen, J., Wang, L., Wang, X., Lu, L., Han, P., Zhang, C., Han, M., Xiang, S., Wang, H., Xuan, L., Li, Z., Lin, H., Nie, X., & Wu, Y. (2025). Genome-Wide Identification and Analysis of the MYC Gene Family in Cotton: Evolution and Expression Profiles During Normal Growth and Stress Response. Genes, 16(1), 20. https://doi.org/10.3390/genes16010020