
Knockout Mouse Studies Show That Mitochondrial CLPP Peptidase and CLPX Unfoldase Act in Matrix Condensates near IMM, as Fast Stress Response in Protein Assemblies for Transcript Processing, Translation, and Heme Production

{kind=link}
Abstract
1. CLPP Is a Key Modifier of Growth and Lifespan, but Its Substrates Remain Unclear
2. Novel Evidence on CLPP and CLPX’s Functions from Clpp-KO Mice and PRLTS3 Patients
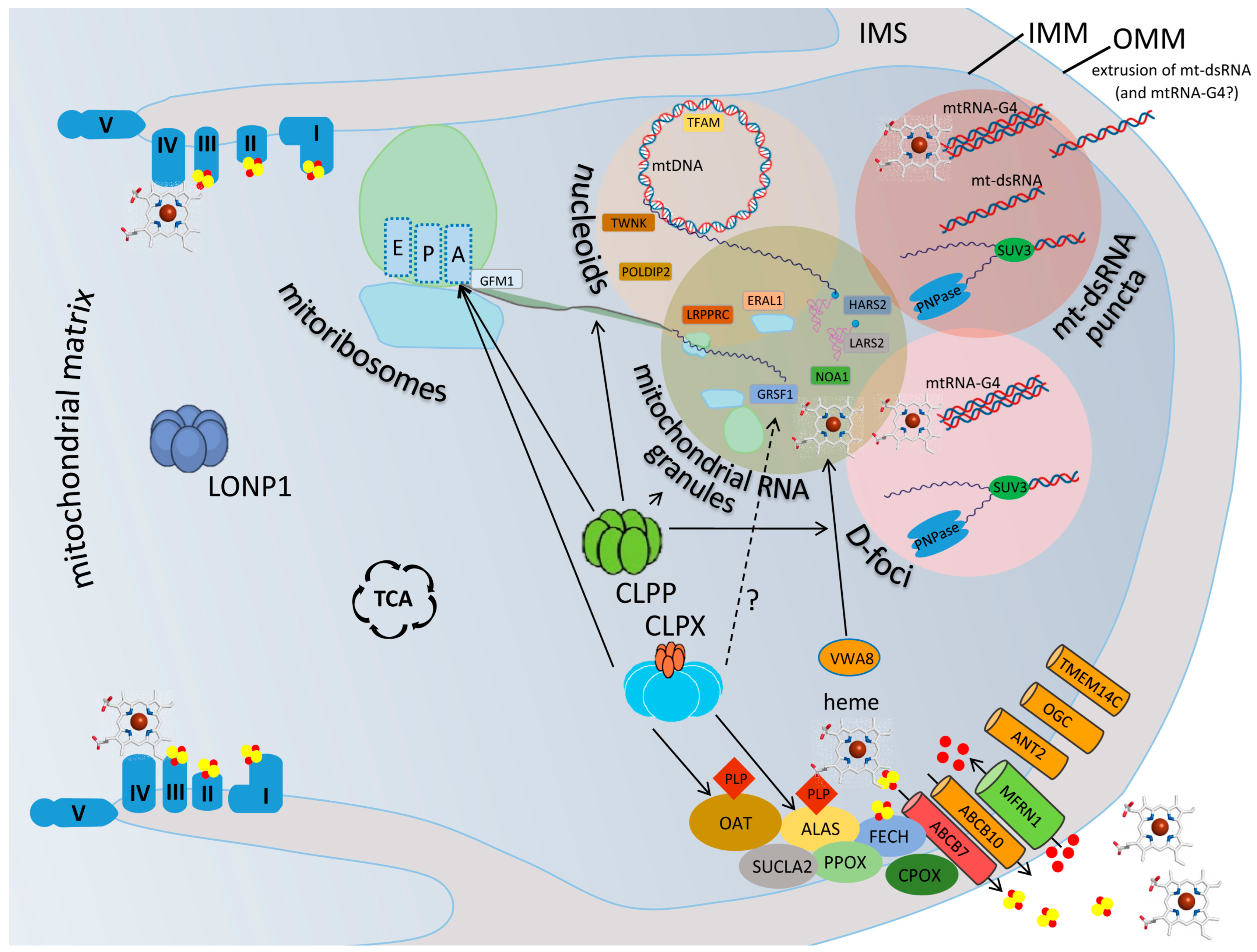
2.1. Prominent Impact of Absence of CLPP and Excess of CLPX on Mitochondrial Nucleoids
2.2. Prominent Impact of Absence of CLPP and Excess of CLPX on Mitoribosomes, Mostly on tRNA-/mRNA-Associated and rRNA-Containing mtSSU
2.3. Prominent Impact of Absence of CLPP and Excess of CLPX on Mitochondrial RNA Processing Granules
2.4. Prominent Impact of Absence of CLPP and Excess of CLPX on Mitochondrial D-Foci Where RNA Degradation, Extrusion, and Innate Immunity Activation Are Decided
2.5. Prominent Impact of Absence of CLPP via CLPX Accumulation on Heme Biosynthesis and Incorporation into Complex-IV of the Respiratory Chain
2.6. Prominent Impact of Absence of CLPP on Fe-S Cluster Containing Peripheral Arm of Respiratory Complex-I
3. Phase-Separated Condensates in Mitochondria and the Cytosol
4. Proposal
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4Fe–4S | iron–sulfur clusters consisting of two interleaved 4Fe- and 4S-tetrahedra |
| AAA+ | ATPases Associated with diverse cellular Activities and other ring-shaped P-loop NTPases |
| ABCB7 | ATP Binding Cassette Subfamily B Member 7 |
| ABCB10 | ATP Binding Cassette Subfamily B Member 10 |
| ALAS | delta-Amino-Levulinic Acid Synthase |
| ALAS2 | delta-Amino-Levulinic Acid Synthase 2, erythroid-specific |
| ALDH18A1 | Aldehyde Dehydrogenase 18 family member A1, =P5CS, delta-1-Pyrroline-5-Carboxylate Synthase |
| ANT2 | Adenine Nucleotide Translocator 2, =SLC25A25 |
| ATP | Adenosine Tri-Phosphate |
| ATPase | Adenosine Tri-Phosphatase |
| CAT-tail | C-terminal alanine and threonine tail |
| cGAS | cyclic GMP-AMP Synthase |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CLPX | Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X |
| CODAS | multiple anomalies syndrome with Cerebral, Ocular, Dental, Auricular and Skeletal anomalies |
| Cox1 | mitochondrially encoded Cytochrome C Oxidase I, mRNA |
| COX15 | Cytochrome C Oxidase assembly homolog COX15 |
| CPOX | Copro-Porphyrinogen OXidase |
| D-foci | degradosome granules in mitochondrial matrix |
| D-loop | displacement loop within the mtDNA |
| DARS2 | Aspartyl-tRNA Synthetase 2, mitochondrial |
| deltaALA | delta-aminolevulinic acid |
| DNA | Deoxyribo-Nucleic Acid |
| DNAzyme | catalytically active DNA sequences |
| dsRNA | double-stranded RNA |
| EF-G | Elongation Factor G |
| EFG1 | G Elongation Factor, mitochondrial 1 |
| ERAL1 | Era (E. coli) -Like 12S mitochondrial rRNA chaperone 1 |
| Fe2+ | divalent cation of iron, ferrous iron |
| FECH | Ferrochelatase |
| Fe-S | iron–sulfur |
| G4 | Guanine quadruplex where RNA or DNA acquires four-stranded conformation |
| GFM1 | G elongation Factor Mitochondrial 1 |
| GRSF1 | G-rich RNA Sequence-binding Factor 1 |
| GSA | Glutamate-5-Semi-Aldehyde |
| GTP | Guanosine-5′-triphosphate |
| HARS2 | Histidyl-tRNA Synthetase 2, mitochondrial |
| hsp-6 | heat shock protein family B (small) member 6 |
| hsp-60 | heat shock protein family D (hsp60) member 1, =human HSPD1 |
| HSPD1 | heat shock protein family D (hsp60) member 1, =human HSP60, chaperonin |
| HTRA2 | High-Temperature-Requirement A serine peptidase 2 |
| i-AAA | ATP-dependent zinc metalloprotease YME1 (S. cerevisiae) -Like 1 |
| IDR | Intrinsically Disordered Region |
| IMM | Inner Membrane of Mitochondria |
| ISC | iron–sulfur cluster |
| KO | knockout |
| LARS2 | Leucyl-tRNA Synthetase 2, mitochondrial |
| lncRNA | long non-coding RNA |
| LLPS | liquid–liquid phase separation |
| LONP1 | Lon Peptidase 1, Mitochondrial |
| LRPPRC | Leucine-Rich Pentatrico-Peptide Repeat Containing |
| m-AAA | AFG3-like matrix AAA peptidase subunit 2 and SPG7 matrix AAA peptidase subunit paraplegin |
| MEFs | mouse embryonic fibroblasts |
| MFRN1 | mitoferrin 1, =SLC25A37, Solute Carrier Family 25 Member 37 |
| Mg2+ | divalent cation of magnesium |
| MPP | mitochondrial processing peptidase |
| MRPL12 | Mitochondrial Ribosomal Protein L12 |
| MRPL18 | Mitochondrial Ribosomal Protein L18 |
| MRPL37 | Mitochondrial Ribosomal Protein L37 |
| MRPL38 | Mitochondrial Ribosomal Protein L38 |
| MRPS15 | Mitochondrial Ribosomal Protein S15 |
| MRPS35 | Mitochondrial Ribosomal Protein S35 |
| mtDNA | mitochondrial DNA |
| mtHSP75 | mitochondrial heat shock protein 75, =human TRAP1 |
| mtLSU | mitoribosomal 39S large subunit |
| mtRNA | mitochondrial RNA |
| mtSSB | mitochondrial single-stranded DNA binding protein |
| mtSSU | mitoribosomal 28S small subunit |
| mt-tRNA | mitochondrial transfer RNA |
| NADH | Nicotinamide Adenine Dinucleotide, reduced form |
| NDUFS2 | NADH:Ubiquinone Oxidoreductase Core Subunit S2 |
| NDUFV1 | NADH:Ubiquinone Oxidoreductase Core Subunit V1 |
| NDUFV2 | NADH:Ubiquinone Oxidoreductase Core Subunit V2 |
| NOA1 | Nitric Oxide Associated 1 |
| NTPase | Nucleoside-Tri-Phosphatase |
| OAT | Ornithine delta-Amino-Transferase |
| OGC | 2-Oxoglutarate/Malate Carrier protein, mitochondrial |
| OMA1 | Overlapping with the M-AAA protease 1 homolog, zinc metallopeptidase |
| OMM | Outer Membrane of Mitochondria |
| PARL | Presenilin-Associated Rhomboid-Like |
| PcG bodies | polycomb bodies |
| PEO1 | Progressive External Ophthalmoplegia 1 protein = TWNK |
| Phe | phenylalanine |
| PLP | Pyridoxal-5′-Phosphate |
| PLPBP | PLP-binding protein |
| PNPase | Polyribo-Nucleotide Phosphorylase/Nucleotidyl-Transferase 1 = PNPT1 in human |
| PNPT1 | Polyribo-Nucleotide Phosphorylase/Nucleotidyl-Transferase 1 = PNPase |
| POLDIP2 | DNA Polymerase Delta Interacting Protein 2 |
| poly(A) tail | poly(adenine) tail of messenger RNAs |
| poly(I:C) | poly(inosinic:cytidylic) acid |
| PPOX | Proto-Porphyrinogen OXidase |
| PRLTS3 | Perrault Syndrome type 3 |
| PRORP | Protein-Only RNase P catalytic subunit |
| qPCR | quantitative Polymerase Chain Reaction |
| RMND1 | Required for Meiotic Nuclear Division 1 homolog |
| RNA | Ribo-Nucleic Acid |
| RNA-G4 | RNA, guanine-rich, in quadruplex conformation |
| RNase | ribonuclease |
| RNAzyme | catalytically active RNA sequences |
| RNF213 | Ring Finger protein 213 |
| rRNA | ribosomal RNA |
| SFXN4 | Sideroflexin 4 |
| SLC25A37 | Solute Carrier family 25 member 37, Mitoferrin 1 |
| SLC25A38 | Solute Carrier Family 25 Member 38, mitochondrial glycine transporter |
| STING | STimulator of INterferon response cGAMP interactor 1 |
| SUCLA2 | Succinate–CoA Ligase ADP-forming subunit β |
| SUPV3L1 | SUV3, Suv3-like RNA helicase |
| TCA | Tri-Carboxylic Acid cycle, =Krebs cycle |
| TFAM | Transcription Factor A, Mitochondrial |
| TIGM | Texas Institute of Genomic Medicine |
| TMEM14C | transmembrane protein 14C |
| tRNA | transfer RNA |
| TWNK | Twinkle mtDNA helicase, =PEO1 |
| UPR | unfolded protein response |
| UPRmt | unfolded protein response in mitochondria |
| Val | valine |
| VWA8 | von Willebrand Factor A domain containing 8 |
References
- Key, J.; Kohli, A.; Barcena, C.; Lopez-Otin, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1(+/−) Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, H.; Kutejova, E.; Suda, K.; Wolpensinger, B.; Lustig, A.; Schatz, G.; Engel, A.; Suzuki, C.K. Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc. Natl. Acad. Sci. USA 1999, 96, 6787–6790. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 2015, 161, 858–867. [Google Scholar] [CrossRef]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A 2015, 167, 1501–1509. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Faridi, R.; Stratton, P.; Salmeri, N.; Morell, R.J.; Khan, A.A.; Usmani, M.A.; Newman, W.G.; Riazuddin, S.; Friedman, T.B. Homozygous novel truncating variant of CLPP associated with severe Perrault syndrome. Clin. Genet. 2024, 105, 584–586. [Google Scholar] [CrossRef]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 12862. [Google Scholar] [CrossRef]
- Theunissen, T.E.; Szklarczyk, R.; Gerards, M.; Hellebrekers, D.M.; Mulder-Den Hartog, E.N.; Vanoevelen, J.; Kamps, R.; de Koning, B.; Rutledge, S.L.; Schmitt-Mechelke, T.; et al. Specific MRI Abnormalities Reveal Severe Perrault Syndrome due to CLPP Defects. Front. Neurol. 2016, 7, 203. [Google Scholar] [CrossRef]
- Dursun, F.; Mohamoud, H.S.; Karim, N.; Naeem, M.; Jelani, M.; Kirmizibekmez, H. A Novel Missense Mutation in the CLPP Gene Causing Perrault Syndrome Type 3 in a Turkish Family. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.; Urquhart, J.E.; O’Sullivan, J.; Williams, S.G.; Bhaskar, S.S.; Jenkinson, E.M.; Lourenco, C.M.; Heiberg, A.; Pearce, S.H.; Shalev, S.A.; et al. Expanding the genotypic spectrum of Perrault syndrome. Clin. Genet. 2017, 91, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Jelani, M.; Alrayes, N.; Mohamoud, H.S.; Almramhi, M.M.; Anshasi, W.; Ahmed, N.A.; Wang, J.; Nasir, J.; Al-Aama, J.Y. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J. Neurol. Sci. 2015, 353, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews®[Internet]: Seattle, WA, USA, 1993. [Google Scholar]
- Kobe, C.; Kracht, L.W.; Timmermann, L.; Bachmann, J.; Schmidt, M.C. Perrault Syndrome with progressive nervous system involvement. Clin. Nucl. Med. 2008, 33, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, M.E.; Coker, S.B.; Fox, L.A. Neurologic anomalies of Perrault syndrome. Am. J. Med. Genet. 1996, 65, 274–276. [Google Scholar] [CrossRef]
- Linssen, W.H.; Van den Bent, M.J.; Brunner, H.G.; Poels, P.J. Deafness, sensory neuropathy, and ovarian dysgenesis: A new syndrome or a broader spectrum of Perrault syndrome? Am. J. Med. Genet. 1994, 51, 81–82. [Google Scholar] [CrossRef]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2022, 141, 805–819. [Google Scholar] [CrossRef]
- Tucker, E.J.; Rius, R.; Jaillard, S.; Bell, K.; Lamont, P.J.; Travessa, A.; Dupont, J.; Sampaio, L.; Dulon, J.; Vuillaumier-Barrot, S.; et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: A peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum. Genet. 2020, 139, 1325–1343. [Google Scholar] [CrossRef] [PubMed]
- Gotta, F.; Lamp, M.; Geroldi, A.; Trevisan, L.; Origone, P.; Fugazza, G.; Fabbri, S.; Nesti, C.; Rubegni, A.; Morani, F.; et al. A novel mutation of Twinkle in Perrault syndrome: A not rare diagnosis? Ann. Hum. Genet. 2020, 84, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.B.; Rea, A.; Thomas, H.B.; Thompson, K.; Olahova, M.; Maroofian, R.; Zamani, M.; He, L.; Sadeghian, S.; Galehdari, H.; et al. Novel homozygous variants in PRORP expand the genotypic spectrum of combined oxidative phosphorylation deficiency 54. Eur. J. Hum. Genet. 2023, 31, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Chatzispyrou, I.A.; Alders, M.; Guerrero-Castillo, S.; Zapata Perez, R.; Haagmans, M.A.; Mouchiroud, L.; Koster, J.; Ofman, R.; Baas, F.; Waterham, H.R.; et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum. Mol. Genet. 2017, 26, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Rioux, A.V.; Bergeron, N.A.; Riopel, J.; Marcoux, N.; Theriault, C.; Gould, P.V.; Garneau, A.P.; Isenring, P. The ever wider clinical spectrum of RMND1-related disorders and limitedness of phenotype-based classifications. J. Mol. Med. 2023, 101, 1229–1236. [Google Scholar] [CrossRef]
- Neyroud, A.S.; Rudinger-Thirion, J.; Frugier, M.; Riley, L.G.; Bidet, M.; Akloul, L.; Simpson, A.; Gilot, D.; Christodoulou, J.; Ravel, C.; et al. LARS2 variants can present as premature ovarian insufficiency in the absence of overt hearing loss. Eur. J. Hum. Genet. 2023, 31, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Ozieblo, D.; Pazik, J.; Stepniak, I.; Skarzynski, H.; Oldak, M. Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement. Genes 2020, 11, 1060. [Google Scholar] [CrossRef]
- Riley, L.G.; Rudinger-Thirion, J.; Frugier, M.; Wilson, M.; Luig, M.; Alahakoon, T.I.; Nixon, C.Y.; Kirk, E.P.; Roscioli, T.; Lunke, S.; et al. The expanding LARS2 phenotypic spectrum: HLASA, Perrault syndrome with leukodystrophy, and mitochondrial myopathy. Hum. Mutat. 2020, 41, 1425–1434. [Google Scholar] [CrossRef]
- Demain, L.A.M.; Gerkes, E.H.; Smith, R.J.H.; Molina-Ramirez, L.P.; O’Keefe, R.T.; Newman, W.G. A recurrent missense variant in HARS2 results in variable sensorineural hearing loss in three unrelated families. J. Hum. Genet. 2020, 65, 305–311. [Google Scholar] [CrossRef]
- Karstensen, H.G.; Rendtorff, N.D.; Hindbaek, L.S.; Colombo, R.; Stein, A.; Birkebaek, N.H.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; Hojland, A.T.; Petersen, M.B.; et al. Novel HARS2 missense variants identified in individuals with sensorineural hearing impairment and Perrault syndrome. Eur. J. Med. Genet. 2020, 63, 103733. [Google Scholar] [CrossRef]
- Demain, L.A.M.; Antunes, D.; O’Sullivan, J.; Bhaskhar, S.S.; O’Keefe, R.T.; Newman, W.G. A known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects. Clin. Genet. 2018, 94, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Kosaki, R.; Horikawa, R.; Fujii, E.; Kosaki, K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am. J. Med. Genet. A 2018, 176, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.C.; Levy-Lahad, E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013, 92, 614–620. [Google Scholar] [CrossRef]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11, 2370. [Google Scholar] [CrossRef] [PubMed]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortes, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030–2033. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef] [PubMed]
- van der Vorm, L.N.; Paw, B.H. Studying disorders of vertebrate iron and heme metabolism using zebrafish. Methods Cell Biol. 2017, 138, 193–220. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef]
- Seiferling, D.; Szczepanowska, K.; Becker, C.; Senft, K.; Hermans, S.; Maiti, P.; Konig, T.; Kukat, A.; Trifunovic, A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016, 17, 953–964. [Google Scholar] [CrossRef]
- Key, J.; Gispert, S.; Koepf, G.; Steinhoff-Wagner, J.; Reichlmeir, M.; Auburger, G. Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling. Int. J. Mol. Sci. 2023, 24, 17503. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Castano, J.G. A comparative study of the chymotrypsin-like activity of the rat liver multicatalytic proteinase and the ClpP from Escherichia coli. J. Biol. Chem. 1993, 268, 21165–21171. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Wong, K.S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; et al. Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome. Structure 2023, 31, 185–200.e110. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Houry, W.A. Chemical Modulation of Human Mitochondrial ClpP: Potential Application in Cancer Therapeutics. ACS Chem. Biol. 2019, 14, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Gispert, S.; Kandi, A.R.; Heinz, D.; Hamann, A.; Osiewacz, H.D.; Meierhofer, D.; Auburger, G. CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation. Biomolecules 2024, 14, 241. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Gispert, S.; Koornneef, L.; Sleddens-Linkels, E.; Kohli, A.; Torres-Odio, S.; Koepf, G.; Amr, S.; Reichlmeir, M.; Harter, P.N.; et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells 2022, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Kukat, A.; Szczepanowska, K.; Hermans, S.; Senft, K.; Brandscheid, C.P.; Maiti, P.; Trifunovic, A. CLPP deficiency protects against metabolic syndrome but hinders adaptive thermogenesis. EMBO Rep. 2018, 19, e45126. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Hofsetz, E.; Demir, F.; Szczepanowska, K.; Kukat, A.; Kizhakkedathu, J.N.; Trifunovic, A.; Huesgen, P.F. The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol. Cell. Proteom. 2020, 19, 1330–1345. [Google Scholar] [CrossRef]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10, 3354. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005, 138, 673–678. [Google Scholar] [CrossRef]
- Strack, P.R.; Brodie, E.J.; Zhan, H.; Schuenemann, V.J.; Valente, L.J.; Saiyed, T.; Lowth, B.R.; Angley, L.M.; Perugini, M.A.; Zeth, K.; et al. Polymerase delta-interacting protein 38 (PDIP38) modulates the stability and activity of the mitochondrial AAA+ protease CLPXP. Commun. Biol. 2020, 3, 646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, P.; Wei, B.; Chen, L.; Song, X.; Pan, Y.; Li, J.; Gan, J.; Zhang, T.; Yang, C.G. Assessment of the structure-activity relationship and antileukemic activity of diacylpyramide compounds as human ClpP agonists. Eur. J. Med. Chem. 2023, 258, 115577. [Google Scholar] [CrossRef]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef]
- Munson, H.E.; De Simone, L.; Schwaede, A.; Bhatia, A.; Mithal, D.S.; Young, N.; Kuntz, N.; Rao, V.K. Axonal polyneuropathy and ataxia in children: Consider Perrault Syndrome, a case report. BMC Med. Genom. 2023, 16, 278. [Google Scholar] [CrossRef]
- Wei, L.; Hou, L.; Ying, Y.Q.; Luo, X.P. A Novel Missense Mutation in TWNK Gene Causing Perrault Syndrome Type 5 in a Chinese Family and Review of the Literature. Pharmgenom. Pers. Med. 2022, 15, 1–8. [Google Scholar] [CrossRef]
- Chen, Z.; Tang, S.; Li, H.; Xu, X.; Lyu, J. Analysis of TWNK variant in a family affected with Perrault syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2020, 37, 739–742. [Google Scholar] [CrossRef]
- Kume, K.; Morino, H.; Miyamoto, R.; Matsuda, Y.; Ohsawa, R.; Kanaya, Y.; Tada, Y.; Kurashige, T.; Kawakami, H. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: A case report. BMC Med. Genet. 2020, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Fekete, B.; Pentelenyi, K.; Rudas, G.; Gal, A.; Grosz, Z.; Illes, A.; Idris, J.; Csukly, G.; Domonkos, A.; Molnar, M.J. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med. Genet. 2019, 20, 198. [Google Scholar] [CrossRef]
- Dominguez-Ruiz, M.; Garcia-Martinez, A.; Corral-Juan, M.; Perez-Alvarez, A.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Duenas, A.; Menendez-Gonzalez, M.; Del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: Overlap of TWNK-related recessive disorders. J. Transl. Med. 2019, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Oldak, M.; Ozieblo, D.; Pollak, A.; Stepniak, I.; Lazniewski, M.; Lechowicz, U.; Kochanek, K.; Furmanek, M.; Tacikowska, G.; Plewczynski, D.; et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Qin, Y.; Achenbach, J.; Li, C.; Kijek, J.; Spahn, C.M.; Nierhaus, K.H. EF-G and EF4: Translocation and back-translocation on the bacterial ribosome. Nat. Rev. Microbiol. 2014, 12, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.E.; Loveland, A.B.; Gamper, H.B., Jr.; Hou, Y.M.; Demo, G.; Korostelev, A.A. Time-resolved cryo-EM visualizes ribosomal translocation with EF-G and GTP. Nat. Commun. 2021, 12, 7236. [Google Scholar] [CrossRef]
- Koripella, R.K.; Sharma, M.R.; Bhargava, K.; Datta, P.P.; Kaushal, P.S.; Keshavan, P.; Spremulli, L.L.; Banavali, N.K.; Agrawal, R.K. Structures of the human mitochondrial ribosome bound to EF-G1 reveal distinct features of mitochondrial translation elongation. Nat. Commun. 2020, 11, 3830. [Google Scholar] [CrossRef] [PubMed]
- Kummer, E.; Ban, N. Structural insights into mammalian mitochondrial translation elongation catalyzed by mtEFG1. EMBO J. 2020, 39, e104820. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, S.; Kumar, V.; Ero, R.; Gao, Y.G. Structure of EF-G-ribosome complex in a pretranslocation state. Nat. Struct. Mol. Biol. 2013, 20, 1077–1084. [Google Scholar] [CrossRef]
- Moraleda, J.M.; Gonzalez, R.; Alegre, A.; Anta, J.P.; San Miguel, J.F. Bone marrow necrosis and treatment with interferon. J. Clin. Pathol. 1986, 39, 1045. [Google Scholar] [CrossRef] [PubMed]
- Sitron, C.S.; Park, J.H.; Giafaglione, J.M.; Brandman, O. Aggregation of CAT tails blocks their degradation and causes proteotoxicity in S. cerevisiae. PLoS ONE 2020, 15, e0227841. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Tantray, I.; Lim, J.; Chen, S.; Li, Y.; Davis, Z.; Sitron, C.; Dong, J.; Gispert, S.; Auburger, G.; et al. MISTERMINATE Mechanistically Links Mitochondrial Dysfunction with Proteostasis Failure. Mol. Cell 2019, 75, 835–848.e838. [Google Scholar] [CrossRef] [PubMed]
- Lytvynenko, I.; Paternoga, H.; Thrun, A.; Balke, A.; Muller, T.A.; Chiang, C.H.; Nagler, K.; Tsaprailis, G.; Anders, S.; Bischofs, I.; et al. Alanine Tails Signal Proteolysis in Bacterial Ribosome-Associated Quality Control. Cell 2019, 178, 76–90.e22. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S.; Roche, E.; Zhou, Y.; Sauer, R.T. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998, 12, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, C.; Smirnov, A. GTPase Era at the heart of ribosome assembly. Front. Mol. Biosci. 2023, 10, 1263433. [Google Scholar] [CrossRef]
- Dennerlein, S.; Rozanska, A.; Wydro, M.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 2010, 430, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, T.; Ohgaki, K.; Yagi, M.; Aoki, Y.; Sakai, A.; Matsumoto, S.; Kang, D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 2010, 38, 5554–5568. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Oxelmark, E.; Jauniaux, N.; de Pinto, V.; Marchini, A.; Tommasino, M. High levels of the mitochondrial large ribosomal subunit protein 40 prevent loss of mitochondrial DNA in null mmf1 Saccharomyces cerevisiae cells. Yeast 2004, 21, 539–548. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, X.; Coots, R.A.; Conn, C.S.; Liu, B.; Qian, S.B. Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat. Struct. Mol. Biol. 2015, 22, 404–410. [Google Scholar] [CrossRef]
- Xu, P.; Wang, L.; Peng, H.; Liu, H.; Liu, H.; Yuan, Q.; Lin, Y.; Xu, J.; Pang, X.; Wu, H.; et al. Disruption of Hars2 in Cochlear Hair Cells Causes Progressive Mitochondrial Dysfunction and Hearing Loss in Mice. Front. Cell. Neurosci. 2021, 15, 804345. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, X.; Meng, F.; Cui, L.; Yi, Q.; Zhao, Q.; Cang, X.; Cai, Z.; Mo, J.Q.; Liang, Y.; et al. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNA(His) mutation. J. Biol. Chem. 2020, 295, 940–954. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Bugiani, M.; Mendes, M.I.; Riley, L.G.; Smith, D.E.C.; Rudinger-Thirion, J.; Frugier, M.; Breur, M.; Crawford, J.; van Gaalen, J.; et al. Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology 2019, 92, e1225–e1237. [Google Scholar] [CrossRef] [PubMed]
- Perli, E.; Fiorillo, A.; Giordano, C.; Pisano, A.; Montanari, A.; Grazioli, P.; Campese, A.F.; Di Micco, P.; Tuppen, H.A.; Genovese, I.; et al. Short peptides from leucyl-tRNA synthetase rescue disease-causing mitochondrial tRNA point mutations. Hum. Mol. Genet. 2016, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Hornig-Do, H.T.; Montanari, A.; Rozanska, A.; Tuppen, H.A.; Almalki, A.A.; Abg-Kamaludin, D.P.; Frontali, L.; Francisci, S.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol. Med. 2014, 6, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Levinger, L.; Morl, M.; Florentz, C. Mitochondrial tRNA 3′ end metabolism and human disease. Nucleic Acids Res. 2004, 32, 5430–5441. [Google Scholar] [CrossRef] [PubMed]
- Rumyantseva, A.; Popovic, M.; Trifunovic, A. CLPP deficiency ameliorates neurodegeneration caused by impaired mitochondrial protein synthesis. Brain 2022, 145, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Lyu, B.; Song, Q. The intricate relationship of G-Quadruplexes and bacterial pathogenicity islands. eLife 2024, 12, RP91985. [Google Scholar] [CrossRef] [PubMed]
- Mestre-Fos, S.; Ito, C.; Moore, C.M.; Reddi, A.R.; Williams, L.D. Human ribosomal G-quadruplexes regulate heme bioavailability. J. Biol. Chem. 2020, 295, 14855–14865. [Google Scholar] [CrossRef]
- Chang, T.; Liu, X.; Cheng, X.; Qi, C.; Mei, H.; Shangguan, D. Selective isolation of G-quadruplexes by affinity chromatography. J. Chromatogr. A 2012, 1246, 62–68. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Liu, Z.; Lin, B.; Yi, H.; Xu, F.; Nie, Z.; Yao, S. Insight into G-quadruplex-hemin DNAzyme/RNAzyme: Adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Res. 2016, 44, 7373–7384. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.C.; Shumayrikh, N.; Sen, D. G-quadruplex structures formed by expanded hexanucleotide repeat RNA and DNA from the neurodegenerative disease-linked C9orf72 gene efficiently sequester and activate heme. PLoS ONE 2014, 9, e106449. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yin, Z.; Xiao, R.; Huang, B.; Cui, Y.; Wang, H.; Xiang, Y.; Wang, L.; Lei, L.; Ye, J.; et al. G-quadruplexes sense natural porphyrin metabolites for regulation of gene transcription and chromatin landscapes. Genome Biol. 2022, 23, 259. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Cuesta, S.M.; Herdy, B.; Abdullah, U.B.; Tannahill, D.; Balasubramanian, S. RNA G-quadruplex structures control ribosomal protein production. Sci. Rep. 2021, 11, 22735. [Google Scholar] [CrossRef] [PubMed]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018, 9, 2558. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016, 30, 1224–1239. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.C. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab. 2013, 17, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Hensen, F.; Potter, A.; van Esveld, S.L.; Tarres-Sole, A.; Chakraborty, A.; Sola, M.; Spelbrink, J.N. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res. 2019, 47, 3680–3698. [Google Scholar] [CrossRef] [PubMed]
- Xavier, V.J.; Martinou, J.C. RNA Granules in the Mitochondria and Their Organization under Mitochondrial Stresses. Int. J. Mol. Sci. 2021, 22, 9502. [Google Scholar] [CrossRef]
- Antonicka, H.; Sasarman, F.; Nishimura, T.; Paupe, V.; Shoubridge, E.A. The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab. 2013, 17, 386–398. [Google Scholar] [CrossRef]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Controlling the mitochondrial antisense—Role of the SUV3-PNPase complex and its co-factor GRSF1 in mitochondrial RNA surveillance. Mol. Cell. Oncol. 2018, 5, e1516452. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, J.; Zhu, Y.; Huang, Q.; Gu, Q.; Tian, S.; Ge, J.; Lin, X.; Sha, W. Guanine-rich RNA sequence binding factor 1 regulates neuronal ferroptosis after spinal cord injury in rats via the GPX4 signaling pathway. Brain Res. 2023, 1818, 148497. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, B.; Heydeck, D.; Jahn, D.; Lasse, M.; Sofi, S.; Ufer, C.; Kuhn, H. Male guanine-rich RNA sequence binding factor 1 knockout mice (Grsf1(−/−)) gain less body weight during adolescence and adulthood. Cell Biosci. 2022, 12, 199. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Kim, K.M.; Pandey, P.R.; Noren Hooten, N.; Munk, R.; Kundu, G.; De, S.; Martindale, J.L.; Yang, X.; Evans, M.K.; et al. Loss of RNA-binding protein GRSF1 activates mTOR to elicit a proinflammatory transcriptional program. Nucleic Acids Res. 2019, 47, 2472–2486. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Goffart, S.; Szibor, M.; Wanrooij, S.; Braun, T. Binding to G-quadruplex RNA activates the mitochondrial GTPase NOA1. Biochim. Biophys. Acta 2013, 1833, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Kazak, L.; Wood, S.R.; Mao, C.C.; Fearnley, I.M.; Walker, J.E.; Holt, I.J. Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res. 2012, 40, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Kolanczyk, M.; Pech, M.; Zemojtel, T.; Yamamoto, H.; Mikula, I.; Calvaruso, M.A.; van den Brand, M.; Richter, R.; Fischer, B.; Ritz, A.; et al. NOA1 is an essential GTPase required for mitochondrial protein synthesis. Mol. Biol. Cell 2011, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Kardon, J.R.; Kruger, M.; Szibor, M.; Baker, T.A.; Braun, T. NOA1, a novel ClpXP substrate, takes an unexpected nuclear detour prior to mitochondrial import. PLoS ONE 2014, 9, e103141. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Ma, W.; Sand, Z.; Finlayson, J.; Wang, T.; Brinton, R.D.; Willis, W.T.; Mandarino, L.J. Von Willebrand factor A domain-containing protein 8 (VWA8) localizes to the matrix side of the inner mitochondrial membrane. Biochem. Biophys. Res. Commun. 2020, 521, 158–163. [Google Scholar] [CrossRef]
- Chen, Z.; Suzuki, H.; Kobayashi, Y.; Wang, A.C.; DiMaio, F.; Kawashima, S.A.; Walz, T.; Kapoor, T.M. Structural Insights into Mdn1, an Essential AAA Protein Required for Ribosome Biogenesis. Cell 2018, 175, 822–834.e818. [Google Scholar] [CrossRef]
- Kawashima, S.A.; Chen, Z.; Aoi, Y.; Patgiri, A.; Kobayashi, Y.; Nurse, P.; Kapoor, T.M. Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell 2016, 167, 512–524.e514. [Google Scholar] [CrossRef] [PubMed]
- Prattes, M.; Lo, Y.H.; Bergler, H.; Stanley, R.E. Shaping the Nascent Ribosome: AAA-ATPases in Eukaryotic Ribosome Biogenesis. Biomolecules 2019, 9, 715. [Google Scholar] [CrossRef]
- Linke, R.; Limmer, M.; Juranek, S.A.; Heine, A.; Paeschke, K. The Relevance of G-Quadruplexes for DNA Repair. Int. J. Mol. Sci. 2021, 22, 12599. [Google Scholar] [CrossRef]
- McShane, E.; Couvillion, M.; Ietswaart, R.; Prakash, G.; Smalec, B.M.; Soto, I.; Baxter-Koenigs, A.R.; Choquet, K.; Churchman, L.S. A kinetic dichotomy between mitochondrial and nuclear gene expression processes. Mol. Cell 2024, 84, 1541–1555.e11. [Google Scholar] [CrossRef]
- Honarmand, S.; Shoubridge, E.A. Poly (A) tail length of human mitochondrial mRNAs is tissue-specific and a mutation in LRPPRC results in transcript-specific patterns of deadenylation. Mol. Genet. Metab. Rep. 2020, 25, 100687. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.C.; Hornig-Do, H.T.; Bruni, F.; Chang, J.H.; Jourdain, A.A.; Martinou, J.C.; Falkenberg, M.; Spahr, H.; Larsson, N.G.; Lewis, R.J.; et al. A human mitochondrial poly(A) polymerase mutation reveals the complexities of post-transcriptional mitochondrial gene expression. Hum. Mol. Genet. 2014, 23, 6345–6355. [Google Scholar] [CrossRef]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012, 40, 8033–8047. [Google Scholar] [CrossRef] [PubMed]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Camara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012, 31, 443–456. [Google Scholar] [CrossRef]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.J.; Skrok, M.M.; Golik, P.; Stepien, P.P. Yeast and human mitochondrial helicases. Biochim. Biophys. Acta 2013, 1829, 842–853. [Google Scholar] [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Malecki, M.; Wojcik, M.A.; Stepien, P.P.; Golik, P. RNA degradation in yeast and human mitochondria. Biochim. Biophys. Acta 2012, 1819, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Clemente, P.; Pajak, A.; Laine, I.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. SUV3 helicase is required for correct processing of mitochondrial transcripts. Nucleic Acids Res. 2015, 43, 7398–7413. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Golzarroshan, B.; Lin, C.L.; Agrawal, S.; Tang, W.H.; Wu, C.J.; Yuan, H.S. Dimeric assembly of human Suv3 helicase promotes its RNA unwinding function in mitochondrial RNA degradosome for RNA decay. Protein Sci. 2022, 31, e4312. [Google Scholar] [CrossRef] [PubMed]
- Toompuu, M.; Tuomela, T.; Laine, P.; Paulin, L.; Dufour, E.; Jacobs, H.T. Polyadenylation and degradation of structurally abnormal mitochondrial tRNAs in human cells. Nucleic Acids Res. 2018, 46, 5209–5226. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3′-to-5′ directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef]
- Sarkar, D.; Fisher, P.B. Human polynucleotide phosphorylase (hPNPase old-35): An RNA degradation enzyme with pleiotrophic biological effects. Cell Cycle 2006, 5, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Piazza, F.; Zappone, M.; Sana, M.; Briani, F.; Deho, G. Polynucleotide phosphorylase of Escherichia coli is required for the establishment of bacteriophage P4 immunity. J. Bacteriol. 1996, 178, 5513–5521. [Google Scholar] [CrossRef]
- Chen, P.L. SUV3 Helicase and Mitochondrial Homeostasis. Int. J. Mol. Sci. 2023, 24, 9233. [Google Scholar] [CrossRef]
- Silva, S.; Camino, L.P.; Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, 11024–11029. [Google Scholar] [CrossRef] [PubMed]
- Carzaniga, T.; Sbarufatti, G.; Briani, F.; Deho, G. Polynucleotide phosphorylase is implicated in homologous recombination and DNA repair in Escherichia coli. BMC Microbiol. 2017, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Tarique, M.; Tuteja, R. Rice SUV3 is a bidirectional helicase that binds both DNA and RNA. BMC Plant Biol. 2014, 14, 283. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Bahlo, M.; Pennisi, A.; Jacoupy, M.; Tankard, R.M.; Ewenczyk, C.; Davies, K.C.; Lino-Coulon, P.; Colace, C.; Rafehi, H.; et al. Heterozygous PNPT1 Variants Cause Spinocerebellar Ataxia Type 25. Ann. Neurol. 2022, 92, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M.; Care4Rare Canada, C. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am. J. Med. Genet. A 2018, 176, 2487–2493. [Google Scholar] [CrossRef]
- Matilainen, S.; Carroll, C.J.; Richter, U.; Euro, L.; Pohjanpelto, M.; Paetau, A.; Isohanni, P.; Suomalainen, A. Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum. Mol. Genet. 2017, 26, 3352–3361. [Google Scholar] [CrossRef]
- Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Matsuhashi, T.; Numata-Uematsu, Y.; Uematsu, M.; Fujii, Y.; Murayama, K.; Ohtake, A.; Abe, T.; et al. Novel biallelic mutations in the PNPT1 gene encoding a mitochondrial-RNA-import protein PNPase cause delayed myelination. Clin. Genet. 2018, 93, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Garcia, S.T.; Chandratillake, G.; Bardakjian, T.; Ullah, E.; Wu, D.; Umeda, K.; Lao, R.; Tang, P.L.; Wan, E.; et al. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects. Clin. Genet. 2015, 88, 468–473. [Google Scholar] [CrossRef]
- von Ameln, S.; Wang, G.; Boulouiz, R.; Rutherford, M.A.; Smith, G.M.; Li, Y.; Pogoda, H.M.; Nurnberg, G.; Stiller, B.; Volk, A.E.; et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 2012, 91, 919–927. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Maletzko, A.; Kohli, A.; Gispert, S.; Torres-Odio, S.; Wittig, I.; Heidler, J.; Barcena, C.; Lopez-Otin, C.; Lei, Y.; et al. Loss of mitochondrial ClpP, Lonp1, and Tfam triggers transcriptional induction of Rnf213, a susceptibility factor for moyamoya disease. Neurogenetics 2020, 21, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Maletzko, A.; Key, J.; Wittig, I.; Gispert, S.; Koepf, G.; Canet-Pons, J.; Torres-Odio, S.; West, A.P.; Auburger, G. Increased presence of nuclear DNAJA3 and upregulation of cytosolic STAT1 and of nucleic acid sensors trigger innate immunity in the ClpP-null mouse. Neurogenetics 2021, 22, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of Mitochondrial Protease CLPP Activates Type I IFN Responses through the Mitochondrial DNA-cGAS-STING Signaling Axis. J. Immunol. 2021, 206, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.; Druyan, R.; Getz, G.S.; Rabinowitz, M. Intramitochondrial localization of delta-aminolaevulate synthetase and ferrochelatase in rat liver. Biochem. J. 1969, 114, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.C.; Andrew, T.L.; Karr, S.W.; Dailey, H.A. Organization of the terminal two enzymes of the heme biosynthetic pathway. Orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J. Biol. Chem. 1988, 263, 3835–3839. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Clark, D.S. Phase-separated biomolecular condensates for biocatalysis. Trends Biotechnol. 2024, 42, 496–509. [Google Scholar] [CrossRef]
- Dahmani, I.; Qin, K.; Zhang, Y.; Fernie, A.R. The formation and function of plant metabolons. Plant J. 2023, 114, 1080–1092. [Google Scholar] [CrossRef]
- Kastritis, P.L.; Gavin, A.C. Enzymatic complexes across scales. Essays Biochem. 2018, 62, 501–514. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed]
- Shum, M.; Shintre, C.A.; Althoff, T.; Gutierrez, V.; Segawa, M.; Saxberg, A.D.; Martinez, M.; Adamson, R.; Young, M.R.; Faust, B.; et al. ABCB10 exports mitochondrial biliverdin, driving metabolic maladaptation in obesity. Sci. Transl. Med. 2021, 13, eabd1869. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.A.; Cowan, J.A. Evolution of the human mitochondrial ABCB7 [2Fe-2S](GS)(4) cluster exporter and the molecular mechanism of an E433K disease-causing mutation. Arch. Biochem. Biophys. 2021, 697, 108661. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Fendley, G.A.; Saxberg, A.D.; Zoghbi, M.E. Stimulation of the human mitochondrial transporter ABCB10 by zinc-mesoporphrin. PLoS ONE 2020, 15, e0238754. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Arimura, H.; Fukushige, T.; Minami, K.; Nishizawa, Y.; Tanimoto, A.; Kanekura, T.; Nakagawa, M.; Akiyama, S.; Furukawa, T. Abcb10 role in heme biosynthesis in vivo: Abcb10 knockout in mice causes anemia with protoporphyrin IX and iron accumulation. Mol. Cell. Biol. 2014, 34, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef]
- Clough, C.A.; Pangallo, J.; Sarchi, M.; Ilagan, J.O.; North, K.; Bergantinos, R.; Stolla, M.C.; Naru, J.; Nugent, P.; Kim, E.; et al. Coordinated missplicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant myelodysplastic syndrome. Blood 2022, 139, 2038–2049. [Google Scholar] [CrossRef]
- Harding, C.R.; Sidik, S.M.; Petrova, B.; Gnadig, N.F.; Okombo, J.; Herneisen, A.L.; Ward, K.E.; Markus, B.M.; Boydston, E.A.; Fidock, D.A.; et al. Genetic screens reveal a central role for heme metabolism in artemisinin susceptibility. Nat. Commun. 2020, 11, 4813. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Robledo, R.F.; Schultz, I.J.; Takahashi-Makise, N.; Gwynn, B.; Bauer, D.E.; Dass, A.; Yi, G.; Li, L.; Hildick-Smith, G.J.; et al. TMEM14C is required for erythroid mitochondrial heme metabolism. J. Clin. Investig. 2014, 124, 4294–4304. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009, 10, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE 2008, 3, e3070. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Ohmori, M.; Shinouchi, K.; Tsuboi, Y.; Hirao, S.; Azuma, M.; Watanabe, H.; Okura, I.; Handa, H. Porphyrin accumulation in mitochondria is mediated by 2-oxoglutarate carrier. J. Biol. Chem. 2006, 281, 31729–31735. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef]
- Deery, E.; Schroeder, S.; Lawrence, A.D.; Taylor, S.L.; Seyedarabi, A.; Waterman, J.; Wilson, K.S.; Brown, D.; Geeves, M.A.; Howard, M.J.; et al. An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat. Chem. Biol. 2012, 8, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.W.; Cohen, N.S.; Raijman, L. Channeling of urea cycle intermediates in situ in permeabilized hepatocytes. J. Biol. Chem. 1989, 264, 4038–4044. [Google Scholar] [CrossRef]
- Chen, C.; Hamza, I. Notes from the Underground: Heme Homeostasis in C. elegans. Biomolecules 2023, 13, 1149. [Google Scholar] [CrossRef]
- Chambers, I.G.; Willoughby, M.M.; Hamza, I.; Reddi, A.R. One ring to bring them all and in the darkness bind them: The trafficking of heme without deliverers. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118881. [Google Scholar] [CrossRef]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharmacol. Exp. Ther. 2016, 356, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.F.; Charoenkwan, P.; Srinivasulu, Y.; Vasylenko, T.; Lai, S.C.; Lee, H.C.; Chen, Y.H.; Huang, H.L.; Ho, S.Y. SCMHBP: Prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinform. 2014, 15 (Suppl. S16), S4. [Google Scholar] [CrossRef]
- Whitman, J.C.; Paw, B.H.; Chung, J. The role of ClpX in erythropoietic protoporphyria. Hematol. Transfus. Cell Ther. 2018, 40, 182–188. [Google Scholar] [CrossRef]
- Fouquet, C.; Le Rouzic, M.A.; Leblanc, T.; Fouyssac, F.; Leverger, G.; Hessissen, L.; Marlin, S.; Bourrat, E.; Fahd, M.; Raffoux, E.; et al. Genotype/phenotype correlations of childhood-onset congenital sideroblastic anaemia in a European cohort. Br. J. Haematol. 2019, 187, 530–542. [Google Scholar] [CrossRef]
- Kardon, J.R.; Moroco, J.A.; Engen, J.R.; Baker, T.A. Mitochondrial ClpX activates an essential biosynthetic enzyme through partial unfolding. eLife 2020, 9, e54387. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.L.; Kardon, J.R.; Sauer, R.T.; Baker, T.A. Structure of the Mitochondrial Aminolevulinic Acid Synthase, a Key Heme Biosynthetic Enzyme. Structure 2018, 26, 580–589.e584. [Google Scholar] [CrossRef]
- Huang, L. An experimental study of the principle of electronic root canal measurement. J. Endod. 1987, 13, 60–64. [Google Scholar] [CrossRef]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, D.; Peracchi, A. A subfamily of PLP-dependent enzymes specialized in handling terminal amines. Biochim. Biophys. Acta 2015, 1854, 1200–1211. [Google Scholar] [CrossRef]
- Tarnacka, B.; Jopowicz, A.; Maslinska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Laut, C.L.; Leasure, C.S.; Pi, H.; Carlin, S.M.; Chu, M.L.; Hillebrand, G.H.; Lin, H.K.; Yi, X.I.; Stauff, D.L.; Skaar, E.P. DnaJ and ClpX Are Required for HitRS and HssRS Two-Component System Signaling in Bacillus anthracis. Infect. Immun. 2022, 90, e0056021. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; Friedman, D.B.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Proteomic analyses of iron-responsive, Clp-dependent changes in Staphylococcus aureus. Pathog. Dis. 2015, 73, ftv004. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J. Bacteriol. 2013, 195, 5041–5050. [Google Scholar] [CrossRef] [PubMed]
- Stanne, T.M.; Sjogren, L.L.; Koussevitzky, S.; Clarke, A.K. Identification of new protein substrates for the chloroplast ATP-dependent Clp protease supports its constitutive role in Arabidopsis. Biochem. J. 2009, 417, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Elliott, M.; Elliott, T. Conditional stability of the HemA protein (glutamyl-tRNA reductase) regulates heme biosynthesis in Salmonella typhimurium. J. Bacteriol. 1999, 181, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Tune instead of destroy: How proteolysis keeps OXPHOS in shape. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148365. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-Lopez, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 43. [Google Scholar] [CrossRef]
- Berrisford, J.M.; Baradaran, R.; Sazanov, L.A. Structure of bacterial respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 892–901. [Google Scholar] [CrossRef]
- Friedrich, T. On the mechanism of respiratory complex I. J. Bioenerg. Biomembr. 2014, 46, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Clason, T.; Ruiz, T.; Schagger, H.; Peng, G.; Zickermann, V.; Brandt, U.; Michel, H.; Radermacher, M. The structure of eukaryotic and prokaryotic complex I. J. Struct. Biol. 2010, 169, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Hohne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020, 11, 1643. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Demarest, T.G.; Tian, J.; Croteau, D.L.; Bohr, V.A.; Misteli, T. Self-assembly of multi-component mitochondrial nucleoids via phase separation. EMBO J. 2021, 40, e107165. [Google Scholar] [CrossRef] [PubMed]
- Rey, T.; Zaganelli, S.; Cuillery, E.; Vartholomaiou, E.; Croisier, M.; Martinou, J.C.; Manley, S. Mitochondrial RNA granules are fluid condensates positioned by membrane dynamics. Nat. Cell Biol. 2020, 22, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Guilhas, B.; Walter, J.C.; Rech, J.; David, G.; Walliser, N.O.; Palmeri, J.; Mathieu-Demaziere, C.; Parmeggiani, A.; Bouet, J.Y.; Le Gall, A.; et al. ATP-Driven Separation of Liquid Phase Condensates in Bacteria. Mol. Cell 2020, 79, 293–303.e294. [Google Scholar] [CrossRef] [PubMed]
- Ladouceur, A.M.; Parmar, B.S.; Biedzinski, S.; Wall, J.; Tope, S.G.; Cohn, D.; Kim, A.; Soubry, N.; Reyes-Lamothe, R.; Weber, S.C. Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc. Natl. Acad. Sci. USA 2020, 117, 18540–18549. [Google Scholar] [CrossRef] [PubMed]
- Muthunayake, N.S.; Tomares, D.T.; Childers, W.S.; Schrader, J.M. Phase-separated bacterial ribonucleoprotein bodies organize mRNA decay. Wiley Interdiscip. Rev. RNA 2020, 11, e1599. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Annunziata, O.; Asherie, N.; Lomakin, A.; Pande, J.; Ogun, O.; Benedek, G.B. Effect of polyethylene glycol on the liquid-liquid phase transition in aqueous protein solutions. Proc. Natl. Acad. Sci. USA 2002, 99, 14165–14170. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, S.; Wang, W.; Shi, J.; Stovall, D.B.; Li, D.; Sui, G. Phase-Separated Subcellular Compartmentation and Related Human Diseases. Int. J. Mol. Sci. 2022, 23, 5491. [Google Scholar] [CrossRef] [PubMed]
- Peran, I.; Mittag, T. Molecular structure in biomolecular condensates. Curr. Opin. Struct. Biol. 2020, 60, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Sepehri, A.; Lazaridis, T. Putative Pore Structures of Amyloid beta 25-35 in Lipid Bilayers. Biochemistry 2023, 62, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Fuxreiter, M. Sequence Determinants of the Aggregation of Proteins within Condensates Generated by Liquid-liquid Phase Separation. J. Mol. Biol. 2022, 434, 167201. [Google Scholar] [CrossRef] [PubMed]
- Stockl, M.T.; Zijlstra, N.; Subramaniam, V. alpha-Synuclein oligomers: An amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol. Neurobiol. 2013, 47, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.G.; Kim, J.K. Compartmentalization and metabolic regulation of glycolysis. J. Cell Sci. 2021, 134, jcs258469. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Plokhikh, K.S.; Manuylov, V.D.; Chesnokov, Y.M.; Vasilov, R.G.; Kuznetsova, I.M.; Turoverov, K.K.; Gordeliy, V.I.; Fonin, A.V.; et al. Order wrapped in chaos: On the roles of intrinsically disordered proteins and RNAs in the arrangement of the mitochondrial enzymatic machines. Int. J. Biol. Macromol. 2024, 267, 131455. [Google Scholar] [CrossRef]
- Zurita Rendon, O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol. 2018, 38, e00412-17. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Trifunovic, A. Mitochondrial matrix proteases: Quality control and beyond. FEBS J. 2022, 289, 7128–7146. [Google Scholar] [CrossRef]
- Feng, Y.; Nouri, K.; Schimmer, A.D. Mitochondrial ATP-Dependent Proteases-Biological Function and Potential Anti-Cancer Targets. Cancers 2021, 13, 2020. [Google Scholar] [CrossRef]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Chan, S.S.; Khalimonchuk, O. Mitochondrial protein quality control: The mechanisms guarding mitochondrial health. Antioxid. Redox Signal. 2015, 22, 977–994. [Google Scholar] [CrossRef]
- Xiang, X.; Dai, Z.; Luo, B.; Zhao, N.; Liu, S.; Sui, J.; Huang, J.; Zhou, Y.; Gu, J.; Zhang, J.; et al. Rational Design of a Novel Class of Human ClpP Agonists through a Ring-Opening Strategy with Enhanced Antileukemia Activity. J. Med. Chem. 2024, 67, 6769–6792. [Google Scholar] [CrossRef] [PubMed]
- Wedam, R.; Greer, Y.E.; Wisniewski, D.J.; Weltz, S.; Kundu, M.; Voeller, D.; Lipkowitz, S. Targeting Mitochondria with ClpP Agonists as a Novel Therapeutic Opportunity in Breast Cancer. Cancers 2023, 15, 1936. [Google Scholar] [CrossRef] [PubMed]
- Walther, R.; Westermann, L.M.; Carmali, S.; Jackson, S.E.; Brotz-Oesterhelt, H.; Spring, D.R. Identification of macrocyclic peptides which activate bacterial cylindrical proteases. RSC Med. Chem. 2023, 14, 1186–1191. [Google Scholar] [CrossRef]
- Zhou, L.L.; Zhang, T.; Xue, Y.; Yue, C.; Pan, Y.; Wang, P.; Yang, T.; Li, M.; Zhou, H.; Ding, K.; et al. Selective activator of human ClpP triggers cell cycle arrest to inhibit lung squamous cell carcinoma. Nat. Commun. 2023, 14, 7069. [Google Scholar] [CrossRef]
- Greer, Y.E.; Hernandez, L.; Fennell, E.M.J.; Kundu, M.; Voeller, D.; Chari, R.; Gilbert, S.F.; Gilbert, T.S.K.; Ratnayake, S.; Tang, B.; et al. Mitochondrial Matrix Protease ClpP Agonists Inhibit Cancer Stem Cell Function in Breast Cancer Cells by Disrupting Mitochondrial Homeostasis. Cancer Res. Commun. 2022, 2, 1144–1161. [Google Scholar] [CrossRef]
- Cobongela, S.Z.Z.; Makatini, M.M.; Mdluli, P.S.; Sibuyi, N.R.S. Acyldepsipeptide Analogues: A Future Generation Antibiotics for Tuberculosis Treatment. Pharmaceutics 2022, 14, 1956. [Google Scholar] [CrossRef]
- Morreale, F.E.; Kleine, S.; Leodolter, J.; Junker, S.; Hoi, D.M.; Ovchinnikov, S.; Okun, A.; Kley, J.; Kurzbauer, R.; Junk, L.; et al. BacPROTACs mediate targeted protein degradation in bacteria. Cell 2022, 185, 2338–2353.e2318. [Google Scholar] [CrossRef]
- Schwarz, M.; Hubner, I.; Sieber, S.A. Tailored Phenyl Esters Inhibit ClpXP and Attenuate Staphylococcus aureus alpha-Hemolysin Secretion. ChemBioChem 2022, 23, e202200253. [Google Scholar] [CrossRef] [PubMed]
- Brotz-Oesterhelt, H.; Vorbach, A. Reprogramming of the Caseinolytic Protease by ADEP Antibiotics: Molecular Mechanism, Cellular Consequences, Therapeutic Potential. Front. Mol. Biosci. 2021, 8, 690902. [Google Scholar] [CrossRef] [PubMed]
- Bosc, C.; Saland, E.; Bousard, A.; Gadaud, N.; Sabatier, M.; Cognet, G.; Farge, T.; Boet, E.; Gotanegre, M.; Aroua, N.; et al. Mitochondrial inhibitors circumvent adaptive resistance to venetoclax and cytarabine combination therapy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1204–1223. [Google Scholar] [CrossRef] [PubMed]
- Binepal, G.; Mabanglo, M.F.; Goodreid, J.D.; Leung, E.; Barghash, M.M.; Wong, K.S.; Lin, F.; Cossette, M.; Bansagi, J.; Song, B.; et al. Development of Antibiotics That Dysregulate the Neisserial ClpP Protease. ACS Infect. Dis. 2020, 6, 3224–3236. [Google Scholar] [CrossRef] [PubMed]
- Griffith, E.C.; Zhao, Y.; Singh, A.P.; Conlon, B.P.; Tangallapally, R.; Shadrick, W.R.; Liu, J.; Wallace, M.J.; Yang, L.; Elmore, J.M.; et al. Ureadepsipeptides as ClpP Activators. ACS Infect. Dis. 2019, 5, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Gronauer, T.F.; Mandl, M.M.; Lakemeyer, M.; Hackl, M.W.; Messner, M.; Korotkov, V.S.; Pachmayr, J.; Sieber, S.A. Design and synthesis of tailored human caseinolytic protease P inhibitors. Chem. Commun. 2018, 54, 9833–9836. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Mabanglo, M.F.; Seraphim, T.V.; Mollica, A.; Mao, Y.Q.; Rizzolo, K.; Leung, E.; Moutaoufik, M.T.; Hoell, L.; Phanse, S.; et al. Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem. Biol. 2018, 25, 1017–1030.e1019. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Nouri, K.; Feng, Y.; Schimmer, A.D. Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis. 2020, 11, 841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maurizi, M.R. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim. Biophys. Acta 2016, 1862, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.; Roy, K.K. ClpP Peptidase as a Plausible Target for the Discovery of Novel Antibiotics. Curr. Drug Targets 2024, 25, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Yang, C.G. Structural insights into the evolutionary simplification of human ClpP activators. Structure 2023, 31, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, L.; Thomy, D.; Lakemeyer, M.; Westermann, L.M.; Ortega, J.; Sieber, S.A.; Sass, P.; Brotz-Oesterhelt, H. Antibiotic Acyldepsipeptides Stimulate the Streptomyces Clp-ATPase/ClpP Complex for Accelerated Proteolysis. mBio 2022, 13, e0141322. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.J.; Sychantha, D.; Hobson, C.; Pawlowski, A.C.; Prehna, G.; Wright, G.D. ClpP inhibitors are produced by a widespread family of bacterial gene clusters. Nat. Microbiol. 2022, 7, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Houry, W.A. Recent structural insights into the mechanism of ClpP protease regulation by AAA+ chaperones and small molecules. J. Biol. Chem. 2022, 298, 101781. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e729. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Ripstein, Z.A.; Vahidi, S.; Houry, W.A.; Rubinstein, J.L.; Kay, L.E. A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. eLife 2020, 9, e52158. [Google Scholar] [CrossRef]
- Ghanbarpour, A.; Cohen, S.E.; Fei, X.; Kinman, L.F.; Bell, T.A.; Zhang, J.J.; Baker, T.A.; Davis, J.H.; Sauer, R.T. A closed translocation channel in the substrate-free AAA+ ClpXP protease diminishes rogue degradation. Nat. Commun. 2023, 14, 7281. [Google Scholar] [CrossRef]
- Kim, S.; Fei, X.; Sauer, R.T.; Baker, T.A. AAA+ protease-adaptor structures reveal altered conformations and ring specialization. Nat. Struct. Mol. Biol. 2022, 29, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Fei, X.; Bell, T.A.; Baker, T.A. Structure and function of ClpXP, a AAA+ proteolytic machine powered by probabilistic ATP hydrolysis. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chaudhary, A.K.; Woytash, J.; Inigo, J.R.; Gokhale, A.A.; Bshara, W.; Attwood, K.; Wang, J.; Spernyak, J.A.; Rath, E.; et al. A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60. J. Clin. Investig. 2022, 132, e149906. [Google Scholar] [CrossRef]
- Kenny, T.C.; Germain, D. From discovery of the CHOP axis and targeting ClpP to the identification of additional axes of the UPRmt driven by the estrogen receptor and SIRT3. J. Bioenerg. Biomembr. 2017, 49, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef]
- Dai, C.Y.; Ng, C.C.; Hung, G.C.C.; Kirmes, I.; Hughes, L.A.; Du, Y.; Brosnan, C.A.; Ahier, A.; Hahn, A.; Haynes, C.M.; et al. ATFS-1 counteracts mitochondrial DNA damage by promoting repair over transcription. Nat. Cell Biol. 2023, 25, 1111–1120. [Google Scholar] [CrossRef]
- Haynes, C.M.; Hekimi, S. Mitochondrial dysfunction, aging, and the mitochondrial unfolded protein response in Caenorhabditis elegans. Genetics 2022, 222, iyac160. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Gispert, S.; Auburger, G. Knockout Mouse Studies Show That Mitochondrial CLPP Peptidase and CLPX Unfoldase Act in Matrix Condensates near IMM, as Fast Stress Response in Protein Assemblies for Transcript Processing, Translation, and Heme Production. Genes 2024, 15, 694. https://doi.org/10.3390/genes15060694
Key J, Gispert S, Auburger G. Knockout Mouse Studies Show That Mitochondrial CLPP Peptidase and CLPX Unfoldase Act in Matrix Condensates near IMM, as Fast Stress Response in Protein Assemblies for Transcript Processing, Translation, and Heme Production. Genes. 2024; 15(6):694. https://doi.org/10.3390/genes15060694
Chicago/Turabian StyleKey, Jana, Suzana Gispert, and Georg Auburger. 2024. "Knockout Mouse Studies Show That Mitochondrial CLPP Peptidase and CLPX Unfoldase Act in Matrix Condensates near IMM, as Fast Stress Response in Protein Assemblies for Transcript Processing, Translation, and Heme Production" Genes 15, no. 6: 694. https://doi.org/10.3390/genes15060694
APA StyleKey, J., Gispert, S., & Auburger, G. (2024). Knockout Mouse Studies Show That Mitochondrial CLPP Peptidase and CLPX Unfoldase Act in Matrix Condensates near IMM, as Fast Stress Response in Protein Assemblies for Transcript Processing, Translation, and Heme Production. Genes, 15(6), 694. https://doi.org/10.3390/genes15060694