
Single-Cell Transcriptomic Profiling Identifies Molecular Phenotypes of Newborn Human Lung Cells

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Single-Cell Suspension Preparation
2.3. Human Single-Cell Sequencing
2.4. Mouse Single-Cell Sequencing
2.5. Integrating Human and Mouse Data
2.6. Flow Cytometry
2.7. Estimation of Maturation State of Human Cells (In Terms of Murine Maturity)
(In Terms of Murine Maturation) (Frequency × Age × Weight)PND3 +
(Frequency × Age × Weight)PND7 +
(Frequency × Age × Weight)PND10}
2.8. In Situ Hybridization and Immunostaining
2.9. Statistics
3. Results
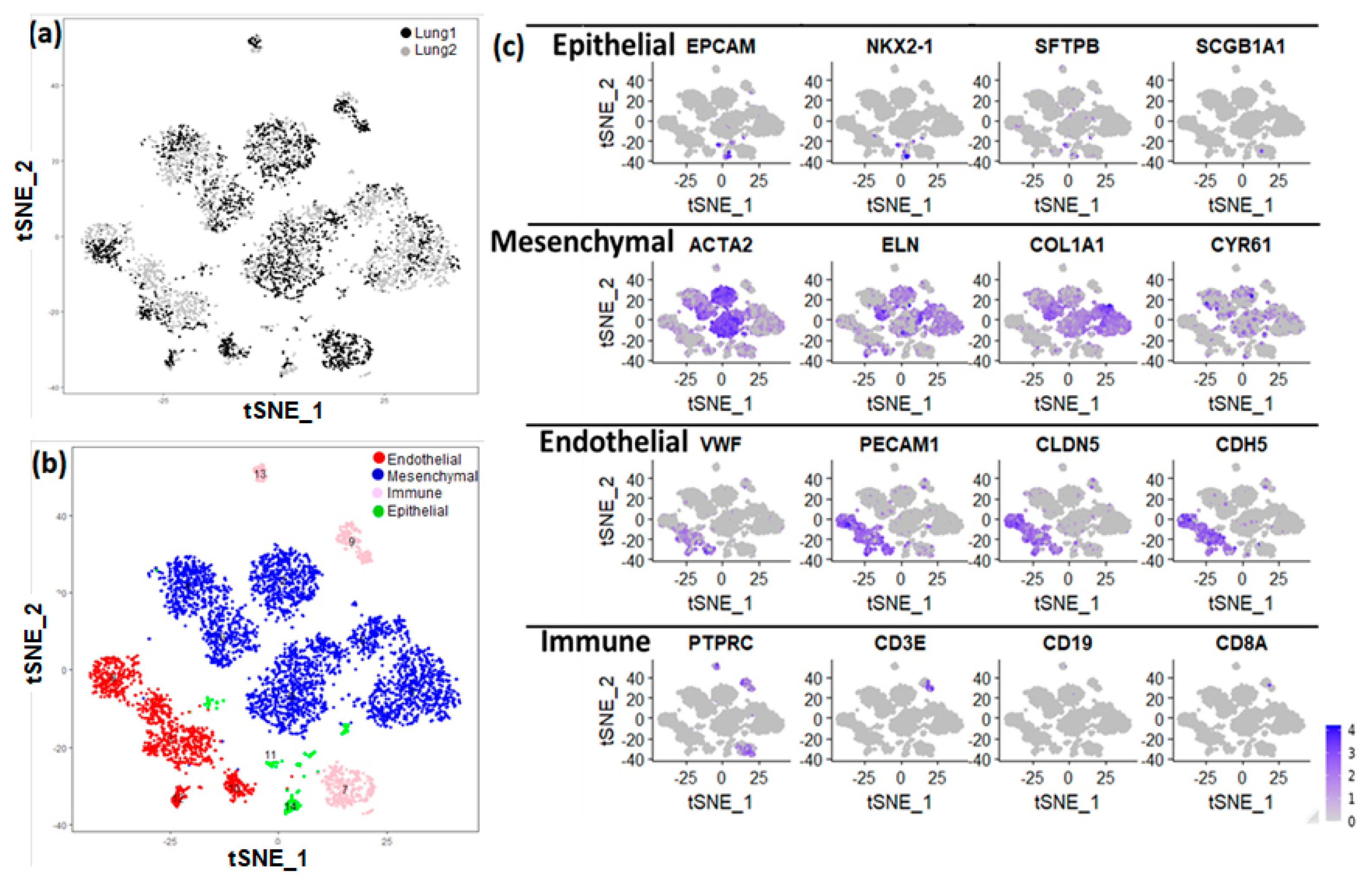
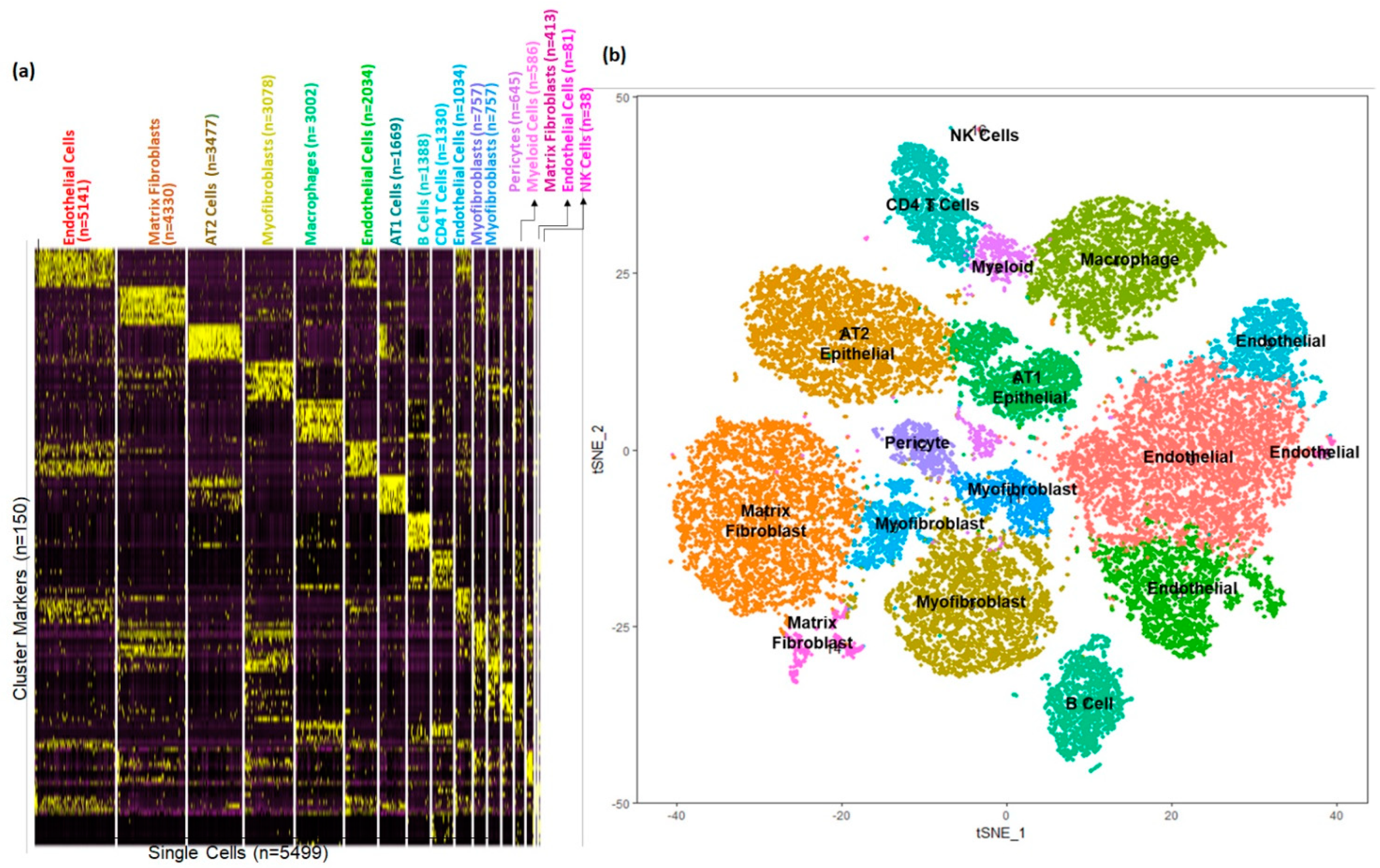
3.1. Cellular Landscape of the Donor Newborn Human Lung
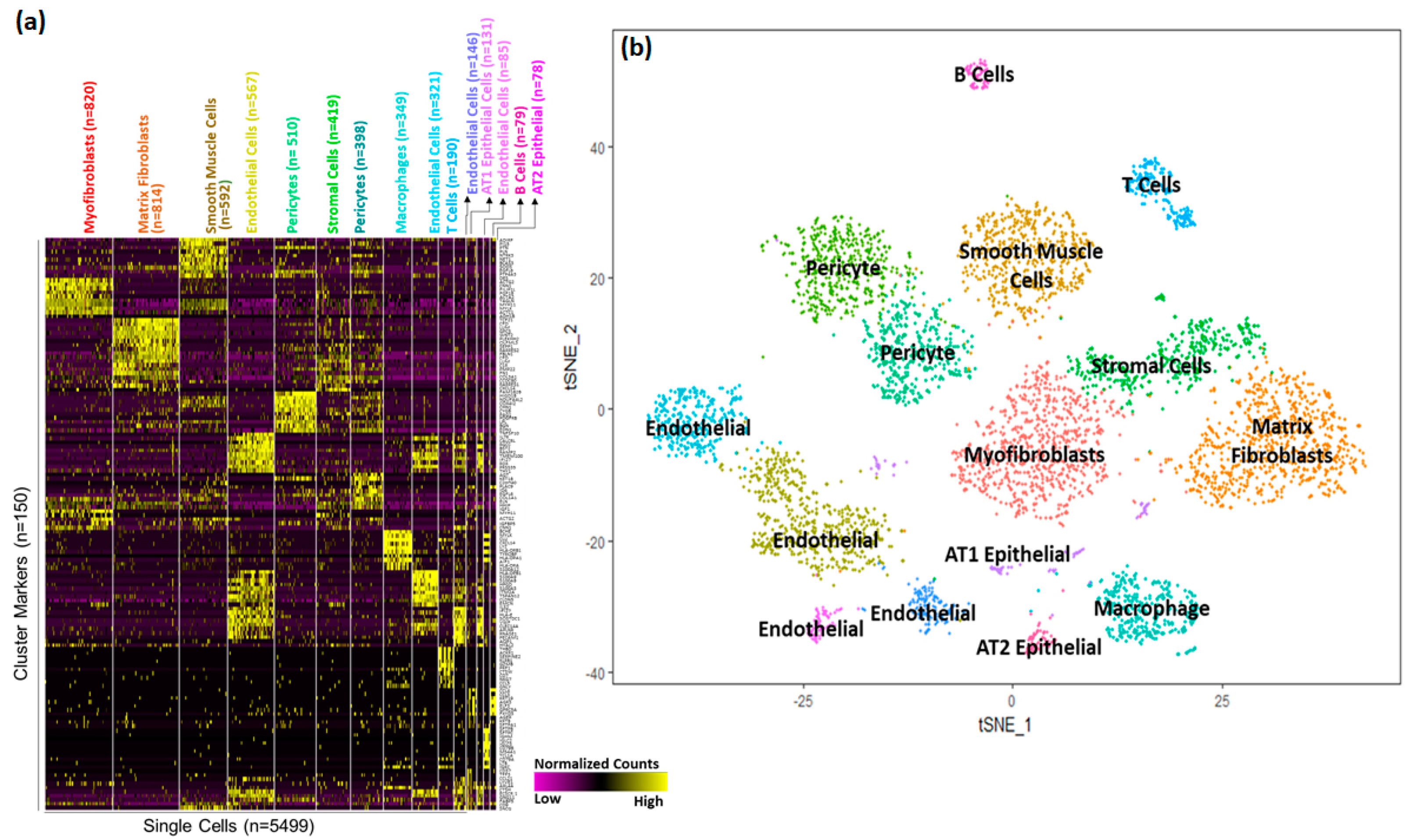
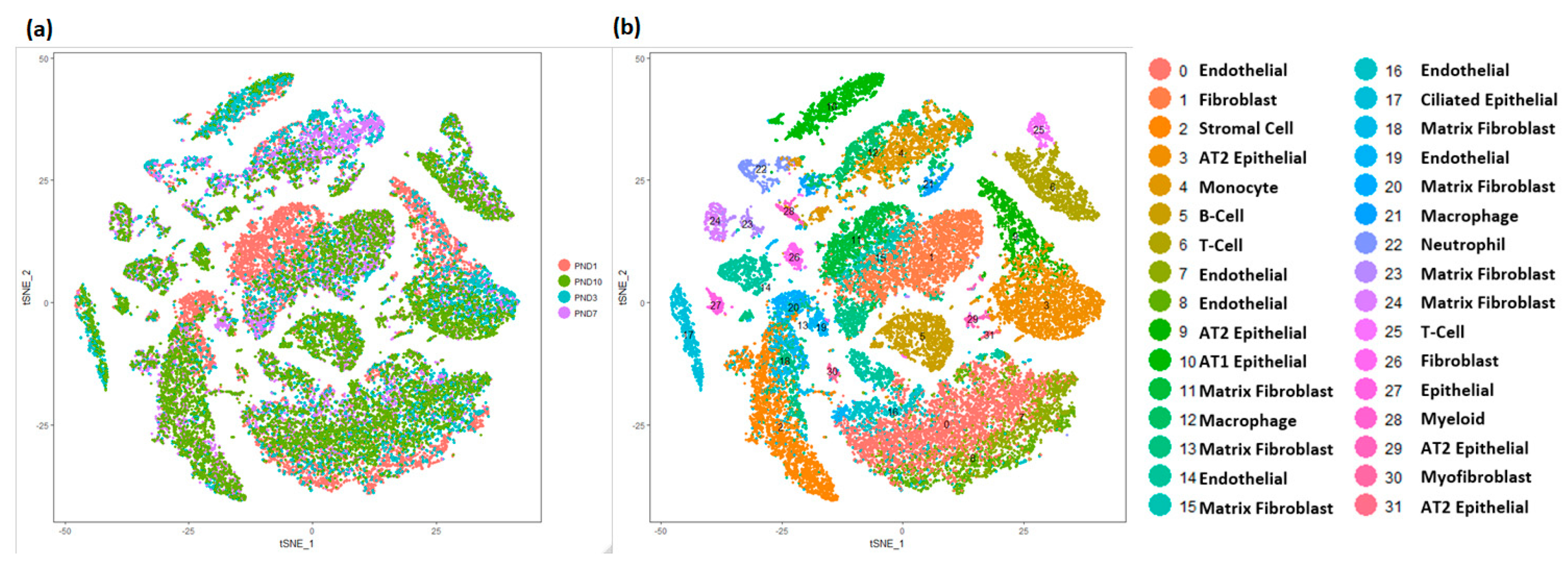
3.2. Cellular Landscape of the Postnatal Mouse Lung
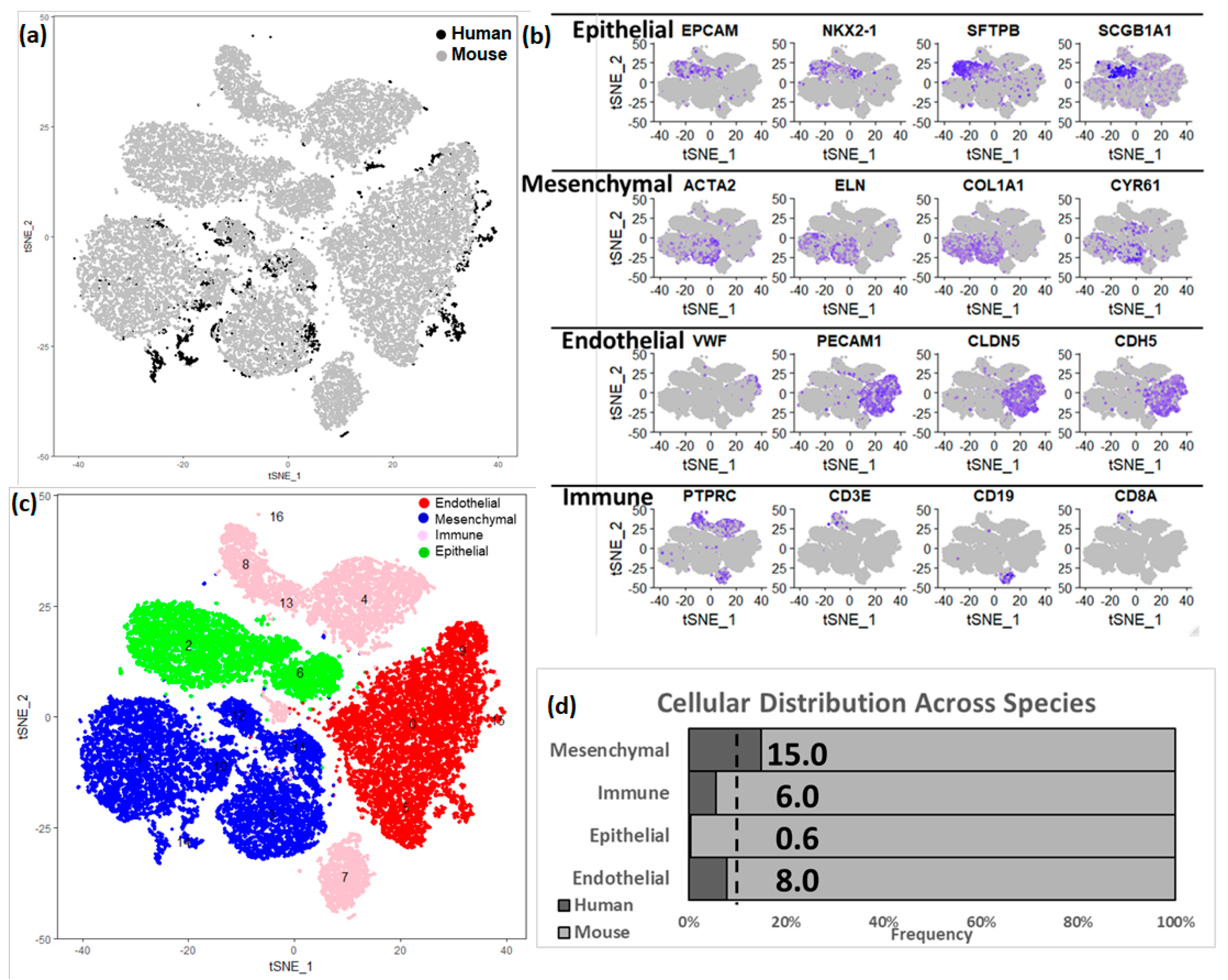
3.3. Integration of Newborn Human and Mouse Lung Data Sets
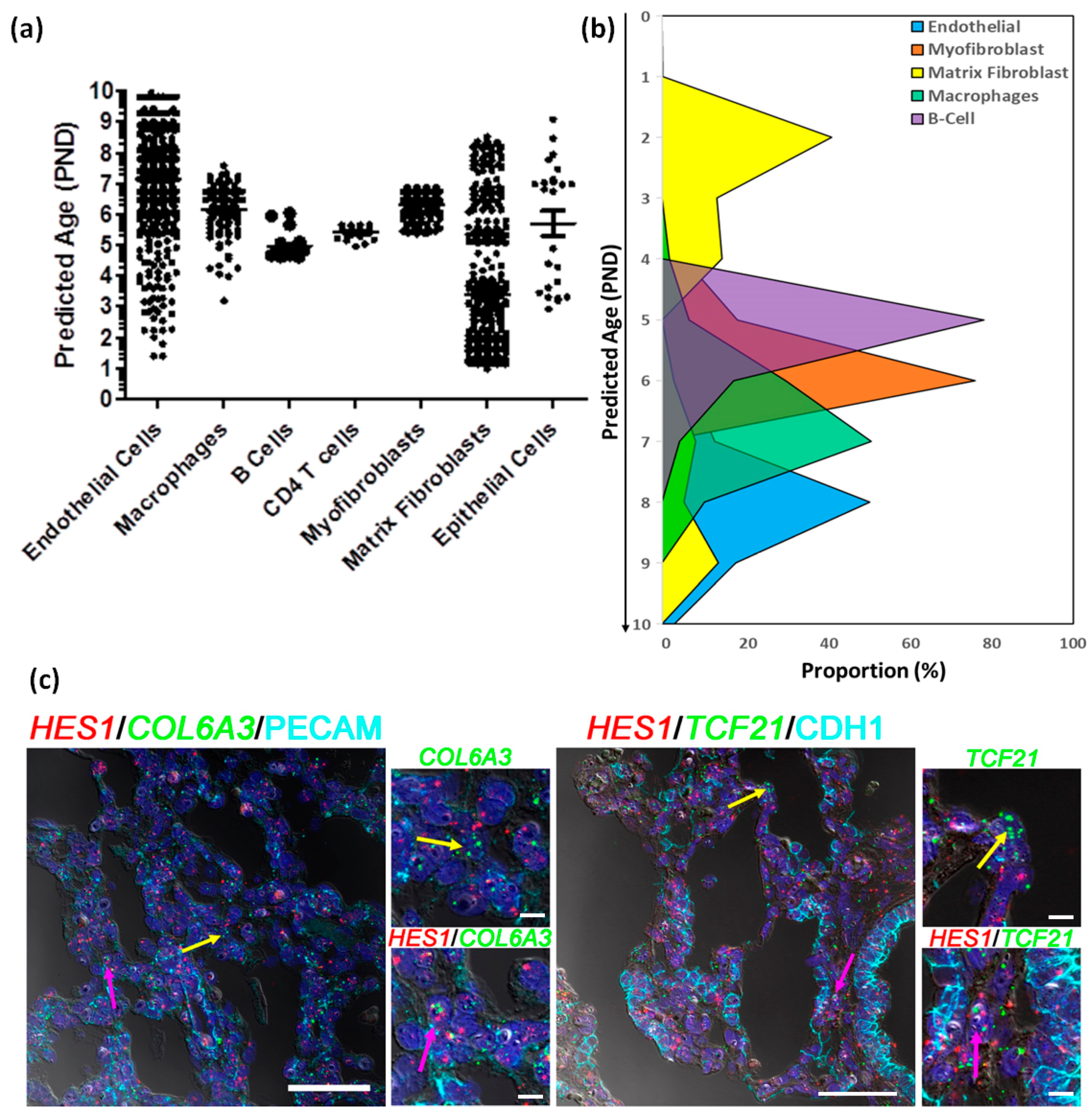
3.4. Estimating Developmental State of Human Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rannels, D.E.; Dunsmore, S.E.; Grove, R.N. Extracellular matrix synthesis and turnover by type II pulmonary epithelial cells. Am. J. Physiol. 1992, 262, L582–L589. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.J.; Colby, T.V.; Travis, W.D.; Tuder, R.M.; Reynolds, H.Y.; Brody, A.R.; Cardoso, W.V.; Crystal, R.G.; Drake, C.J.; Engelhardt, J.; et al. Resident cellular components of the human lung: Current knowledge and goals for research on cell phenotyping and function. Proc. Am. Thorac. Soc. 2008, 5, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Besnard, V.; Wert, S.E.; Ikegami, M.; Xu, Y.; Heffner, C.; Murray, S.A.; Donahue, L.R.; Whitsett, J.A. Maternal synchronization of gestational length and lung maturation. PLoS ONE 2011, 6, e26682. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, K.J.; Wells, J.M.; Kho, A.T.; Philip, V.M.; Kamir, D.; Kohane, I.S.; Graber, J.H.; Bult, C.J. Temporal dynamics of the developing lung transcriptome in three common inbred strains of laboratory mice reveals multiple stages of postnatal alveolar development. PeerJ 2016, 4, e2318. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Y.; Besnard, V.; Ikegami, M.; Wert, S.E.; Heffner, C.; Murray, S.A.; Donahue, L.R.; Whitsett, J.A. Transcriptional programs controlling perinatal lung maturation. PLoS ONE 2012, 7, e37046. [Google Scholar]
- Anderson, C.S.; Chu, C.Y.; Wang, Q.; Mereness, J.A.; Ren, Y.; Donlon, K.; Bhattacharya, S.; Misra, R.S.; Walsh, E.E.; Pryhuber, G.S.; et al. CX3CR1 as a respiratory syncytial virus receptor in pediatric human lung. Pediatr. Res. 2020, 87, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Go, D.; Krenitsky, D.L.; Huyck, H.L.; Solleti, S.K.; Lunger, V.A.; Metlay, L.; Srisuma, S.; Wert, S.E.; Mariani, T.J.; et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2012, 186, 349–358. [Google Scholar] [CrossRef]
- Steiner, L.A.; Getman, M.; Lester, G.M.S.; Iqbal, M.A.; Katzman, P.; Szafranski, P.; Stankiewicz, P.; Bhattacharya, S.; Mariani, T.; Pryhuber, G.; et al. Disruption of normal patterns of FOXF1 expression in a lethal disorder of lung development. J. Med. Genet. 2019, 57, 296–300. [Google Scholar] [CrossRef]
- Wang, Q.; Bhattacharya, S.; Mereness, J.A.; Anderson, C.; Lillis, J.A.; Misra, R.S.; Romas, S.; Huyck, H.; Howell, A.; Bandyopadhyay, G.; et al. A novel in vitro model of primary human pediatric lung epithelial cells. Pediatr. Res. 2020, 87, 511–517. [Google Scholar] [CrossRef]
- Ubags, N.D.J.; Alcazar, M.A.A.; Kallapur, S.G.; Knapp, S.; Lanone, S.; Lloyd, C.M.; Morty, R.E.; Pattaroni, C.; Reynaert, N.L.; Rottier, R.J.; et al. Early origins of lung disease: Towards an interdisciplinary approach. Eur. Respir. Rev. 2020, 29, 200191. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef]
- Besnard, V.; Matsuzaki, Y.; Clark, J.; Xu, Y.; Wert, S.E.; Ikegami, M.; Stahlman, M.T.; Weaver, T.E.; Hunt, A.N.; Postle, A.D.; et al. Conditional deletion of Abca3 in alveolar type II cells alters surfactant homeostasis in newborn and adult mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L646–L659. [Google Scholar] [CrossRef]
- Mariani, T.J.; Reed, J.J.; Shapiro, S.D. Expression profiling of the developing mouse lung: Insights into the establishment of the extracellular matrix. Am. J. Respir. Cell Mol. Biol. 2002, 26, 541–548. [Google Scholar] [CrossRef]
- Kho, A.T.; Bhattacharya, S.; Mecham, B.H.; Hong, J.; Kohane, I.S.; Mariani, T.J. Expression profiles of the mouse lung identify a molecular signature of time-to-birth. Am. J. Respir. Cell Mol. Biol. 2009, 40, 47–57. [Google Scholar] [CrossRef]
- Mereness, J.A.; Bhattacharya, S.; Ren, Y.; Wang, Q.; Anderson, C.S.; Donlon, K.; Dylag, A.M.; Haak, J.; Angelin, A.; Bonaldo, P.; et al. Collagen VI Deficiency Results in Structural Abnormalities in the Mouse Lung. Am. J. Pathol. 2020, 190, 426–441. [Google Scholar] [CrossRef]
- Du, R.; Tantisira, K.; Carey, V.; Bhattacharya, S.; Metje, S.; Kho, A.T.; Klanderman, B.J.; Gaedigk, R.; Lazarus, R.; Mariani, T.J.; et al. Platform dependence of inference on gene-wise and gene-set involvement in human lung development. BMC Bioinform. 2009, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.T.; Bhattacharya, S.; Tantisira, K.G.; Carey, V.J.; Gaedigk, R.; Leeder, J.S.; Kohane, I.S.; Weiss, S.T.; Mariani, T.J. Transcriptomic analysis of human lung development. Am. J. Respir. Crit. Care Med. 2010, 181, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Mariani, T.J. Systems biology approaches to identify developmental bases for lung diseases. Pediatr. Res. 2013, 73, 514–522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Du, Y.; Clair, G.C.; Al Alam, D.; Danopoulos, S.; Schnell, D.; Kitzmiller, J.A.; Misra, R.S.; Bhattacharya, S.; Warburton, D.; Mariani, T.J.; et al. Integration of transcriptomic and proteomic data identifies biological functions in cell populations from human infant lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L347–L360. [Google Scholar] [CrossRef] [PubMed]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef] [PubMed]
- Ardini-Poleske, M.E.; Clark, R.F.; Ansong, C.; Carson, J.P.; Corley, R.A.; Deutsch, G.H.; Hagood, J.S.; Kaminski, N.; Mariani, T.J.; Potter, S.S.; et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L733–L740. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Deutsch, G.H.; Wert, S.E.; Ontology Subcommittee; NHLBI Molecular Atlas of Lung Development Program Consortium. Comprehensive anatomic ontologies for lung development: A comparison of alveolar formation and maturation within mouse and human lung. J. Biomed. Semant. 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Clair, G.; Tsai, C.F.; Xu, K.; Chrisler, W.B.; Sontag, R.L.; Zhao, R.; Moore, R.J.; Liu, T.; Pasa-Tolic, L.; et al. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91, 13119–13127. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gaietto, K.; Chen, W. Mapping a New Course to Understand Lung Biology Mechanisms: LungMAP.net. Am. J. Respir. Cell Mol. Biol. 2024, 70, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef]
- Danopoulos, S.; Bhattacharya, S.; Mariani, T.J.; Al Alam, D. Transcriptional characterisation of human lung cells identifies novel mesenchymal lineage markers. Eur. Respir. J. 2020, 55, 1900746. [Google Scholar] [CrossRef]
- Guo, M.; Du, Y.; Gokey, J.J.; Ray, S.; Bell, S.M.; Adam, M.; Sudha, P.; Perl, A.K.; Deshmukh, H.; Potter, S.S.; et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat. Commun. 2019, 10, 37. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef]
- Schiller, H.B.; Montoro, D.T.; Simon, L.M.; Rawlins, E.L.; Meyer, K.B.; Strunz, M.; Braga, F.A.V.; Timens, W.; Koppelman, G.H.; Budinger, G.R.S.; et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 31–41. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Cohen, M.; Giladi, A.; Gorki, A.-D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.-M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044.e18. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Sikkema, L.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smillie, C.; et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021, 27, 546–559. [Google Scholar] [CrossRef]
- Wang, A.; Chiou, J.; Poirion, O.B.; Buchanan, J.; Valdez, M.J.; Verheyden, J.M.; Hou, X.; Kudtarkar, P.; Narendra, S.; Newsome, J.M.; et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. Elife 2020, 9, e62522. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.; Huyck, H.L.; Misra, R.S.; Bhattacharya, S.; Wang, Q.; Mereness, J.; Lillis, J.; Myers, J.R.; Ashton, J.; Bushnell, T.; et al. Dissociation, cellular isolation, and initial molecular characterization of neonatal and pediatric human lung tissues. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L576–L583. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.E.; Clair, G.; Bandyopadhyay, G.; Misra, R.S.; Zink, E.M.; Bloodsworth, K.J.; Shukla, A.K.; Du, Y.; Lillis, J.; Myers, J.R.; et al. Cell type-resolved human lung lipidome reveals cellular cooperation in lung function. Sci. Rep. 2018, 8, 13455. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323.e30. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Misra, R.S.; Bhattacharya, S.; Huyck, H.L.; Wang, J.C.; Slaunwhite, C.G.; Slaunwhite, S.L.; Wightman, T.R.; Secor-Socha, S.; Misra, S.K.; Bushnell, T.P.; et al. Flow-based sorting of neonatal lymphocyte populations for transcriptomics analysis. J. Immunol. Methods 2016, 437, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, S.; Thornton, M.E.; Grubbs, B.H.; Frey, M.R.; Warburton, D.; Bellusci, S.; Al Alam, D. Discordant roles for FGF ligands in lung branching morphogenesis between human and mouse. J. Pathol. 2018, 247, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Ilicic, T.; Kim, J.K.; Kolodziejczyk, A.A.; Bagger, F.O.; McCarthy, D.J.; Marioni, J.C.; Teichmann, S.A. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Restori, K.H.; Srinivasa, B.T.; Ward, B.J.; Fixman, E.D. Neonatal Immunity, Respiratory Virus Infections, and the Development of Asthma. Front. Immunol. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gillich, A.; Zhang, F.; Farmer, C.G.; Travaglini, K.J.; Tan, S.Y.; Gu, M.; Zhou, B.; Feinstein, J.A.; Krasnow, M.A.; Metzger, R.J. Capillary cell-type specialization in the alveolus. Nature 2020, 586, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Yu, Q.; Czerwinski, M.; Tsai, Y.-H.; Conway, R.F.; Wu, A.; Holloway, E.M.; Walker, T.; Glass, I.A.; Treutlein, B.; et al. In Vitro and In Vivo Development of the Human Airway at Single-Cell Resolution. Dev. Cell 2020, 53, 117–128.e6. [Google Scholar] [CrossRef] [PubMed]
- Sikkema, L.; Ramírez-Suástegui, C.; Strobl, D.C.; Gillett, T.E.; Zappia, L.; Madissoon, E.; Markov, N.S.; Zaragosi, L.-E.; Ji, Y.; Ansari, M.; et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 2023, 29, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Soerens, A.G. Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Rudd, B.D. Neonatal T Cells: A Reinterpretation. Annu. Rev. Immunol. 2020, 38, 229–247. [Google Scholar] [CrossRef]
- Haddad, M.E.; Karlmark, K.; Donato, X.-C.; Martin, G.; Bretelle, F.; Lesavre, N.; Cocallemen, J.-F.; Martin, M.; Picard, C.; Roudier, J.; et al. Corrigendum: Factors Predicting the Presence of Maternal Cells in Cord Blood and Associated Changes in Immune Cell Composition. Front. Immunol. 2021, 12, 763236. [Google Scholar] [CrossRef]
- Scaffa, A.; Yao, H.; Oulhen, N.; Wallace, J.; Peterson, A.L.; Rizal, S.; Ragavendran, A.; Wessel, G.; De Paepe, M.E.; Dennery, P.A. Single-cell transcriptomics reveals lasting changes in the lung cellular landscape into adulthood after neonatal hyperoxic exposure. Redox Biol. 2021, 48, 102091. [Google Scholar] [CrossRef]
- Anderson, K.G.; Mayer-Barber, K.; Sung, H.; Beura, L.; James, B.R.; Taylor, J.J.; Qunaj, L.; Griffith, T.S.; Vezys, V.; Barber, D.L.; et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014, 9, 209–222. [Google Scholar] [CrossRef]
- Adam, M.; Potter, A.S.; Potter, S.S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development 2017, 144, 3625–3632. [Google Scholar] [CrossRef]
- Quatromoni, J.G.; Singhal, S.; Bhojnagarwala, P.; Hancock, W.W.; Albelda, S.M.; Eruslanov, E. An optimized disaggregation method for human lung tumors that preserves the phenotype and function of the immune cells. J. Leukoc. Biol. 2015, 97, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Madissoon, E.; Wilbrey-Clark, A.; Miragaia, R.J.; Saeb-Parsy, K.; Mahbubani, K.T.; Georgakopoulos, N.; Harding, P.; Polanski, K.; Huang, N.; Nowicki-Osuch, K.; et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.J.; Budinger, G.R.S.; Reyfman, P.A. Breathing fresh air into respiratory research with single-cell RNA sequencing. Eur. Respir. Rev. 2020, 29, 200060. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef]
- Frank, D.B.; Penkala, I.J.; Zepp, J.A.; Sivakumar, A.; Linares-Saldana, R.; Zacharias, W.J.; Stolz, K.G.; Pankin, J.; Lu, M.; Wang, Q.; et al. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc. Natl. Acad. Sci. USA 2019, 116, 4362–4371. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Niethamer, T.K.; Stabler, C.T.; Leach, J.P.; Zepp, J.A.; Morley, M.P.; Babu, A.; Zhou, S.; Morrisey, E.E. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife 2020, 9, e53072. [Google Scholar] [CrossRef]
- Xie, T.; Wang, Y.; Deng, N.; Huang, G.; Taghavifar, F.; Geng, Y.; Liu, N.; Kulur, V.; Yao, C.; Chen, P.; et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep. 2018, 22, 3625–3640. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Ou-Yang, H.F.; Wu, C.G.; Qu, S.Y.; Xu, X.T.; Wang, P. Notch signaling regulates col1alpha1 and col1alpha2 expression in airway fibroblasts. Exp. Biol. Med. 2014, 239, 1589–1596. [Google Scholar] [CrossRef]
- Liu, T.; Hu, B.; Choi, Y.Y.; Chung, M.; Ullenbruch, M.; Yu, H.; Lowe, J.B.; Phan, S.H. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am. J. Pathol. 2009, 174, 1745–1755. [Google Scholar] [CrossRef]
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yu, H.; Wei, W.; Cheng, Y.; Huang, S.; Shi, H.; Liu, S.; Xia, J.; Jia, H.; Hao, L. Linkage disequilibrium and functional analysis of PRE1 insertion together with SNPs in the promoter region of IGFBP7 gene in different pig breeds. J. Appl. Genet. 2018, 59, 231–241. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, S.; Myers, J.A.; Baker, C.; Guo, M.; Danopoulos, S.; Myers, J.R.; Bandyopadhyay, G.; Romas, S.T.; Huyck, H.L.; Misra, R.S.; et al. Single-Cell Transcriptomic Profiling Identifies Molecular Phenotypes of Newborn Human Lung Cells. Genes 2024, 15, 298. https://doi.org/10.3390/genes15030298
Bhattacharya S, Myers JA, Baker C, Guo M, Danopoulos S, Myers JR, Bandyopadhyay G, Romas ST, Huyck HL, Misra RS, et al. Single-Cell Transcriptomic Profiling Identifies Molecular Phenotypes of Newborn Human Lung Cells. Genes. 2024; 15(3):298. https://doi.org/10.3390/genes15030298
Chicago/Turabian StyleBhattacharya, Soumyaroop, Jacquelyn A. Myers, Cameron Baker, Minzhe Guo, Soula Danopoulos, Jason R. Myers, Gautam Bandyopadhyay, Stephen T. Romas, Heidie L. Huyck, Ravi S. Misra, and et al. 2024. "Single-Cell Transcriptomic Profiling Identifies Molecular Phenotypes of Newborn Human Lung Cells" Genes 15, no. 3: 298. https://doi.org/10.3390/genes15030298
APA StyleBhattacharya, S., Myers, J. A., Baker, C., Guo, M., Danopoulos, S., Myers, J. R., Bandyopadhyay, G., Romas, S. T., Huyck, H. L., Misra, R. S., Dutra, J., Holden-Wiltse, J., McDavid, A. N., Ashton, J. M., Al Alam, D., Potter, S. S., Whitsett, J. A., Xu, Y., Pryhuber, G. S., & Mariani, T. J. (2024). Single-Cell Transcriptomic Profiling Identifies Molecular Phenotypes of Newborn Human Lung Cells. Genes, 15(3), 298. https://doi.org/10.3390/genes15030298