
Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis

Abstract
1. Introduction
2. Materials and Methods
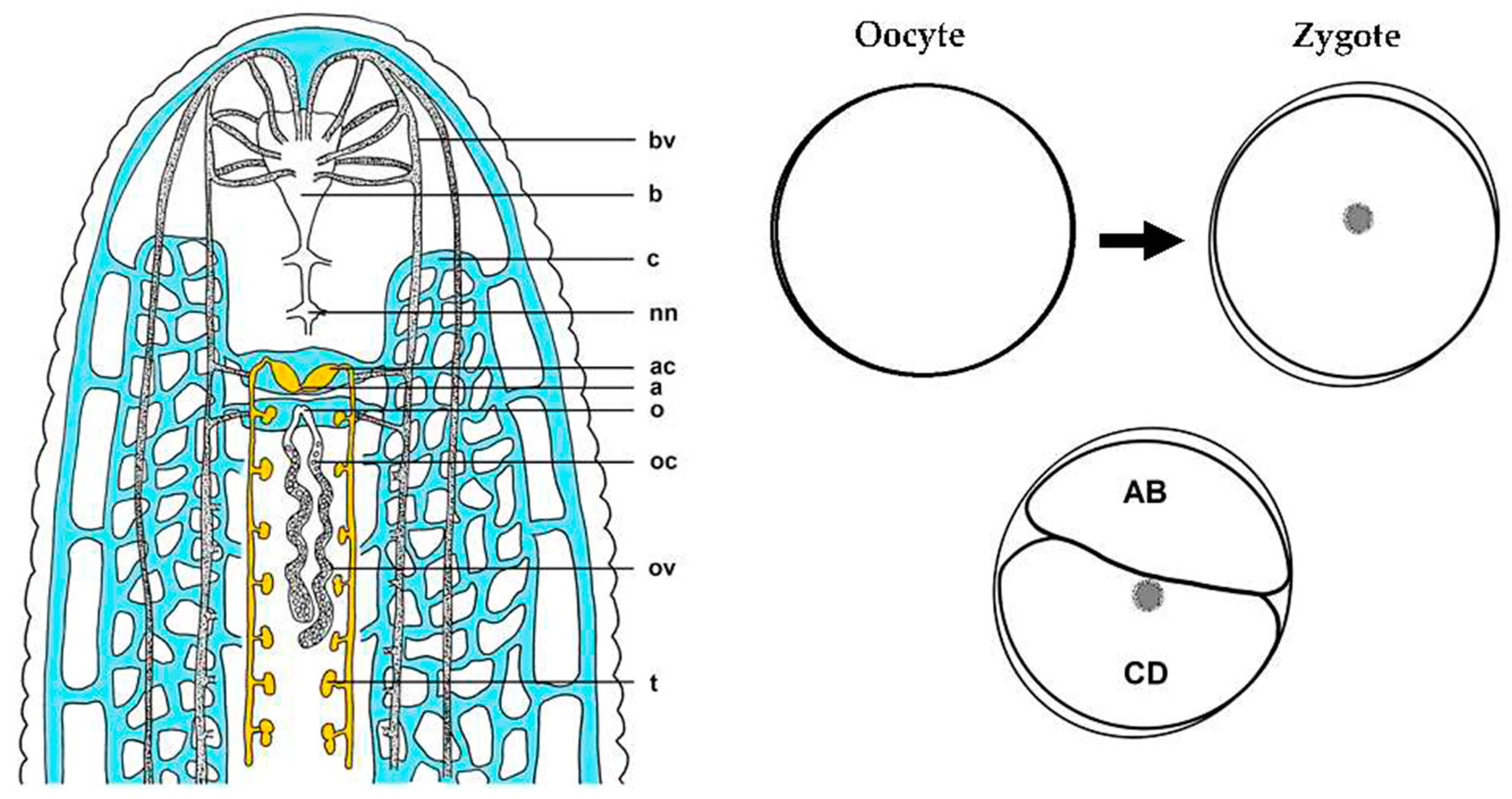
2.1. Specimens and Cell Acquisition
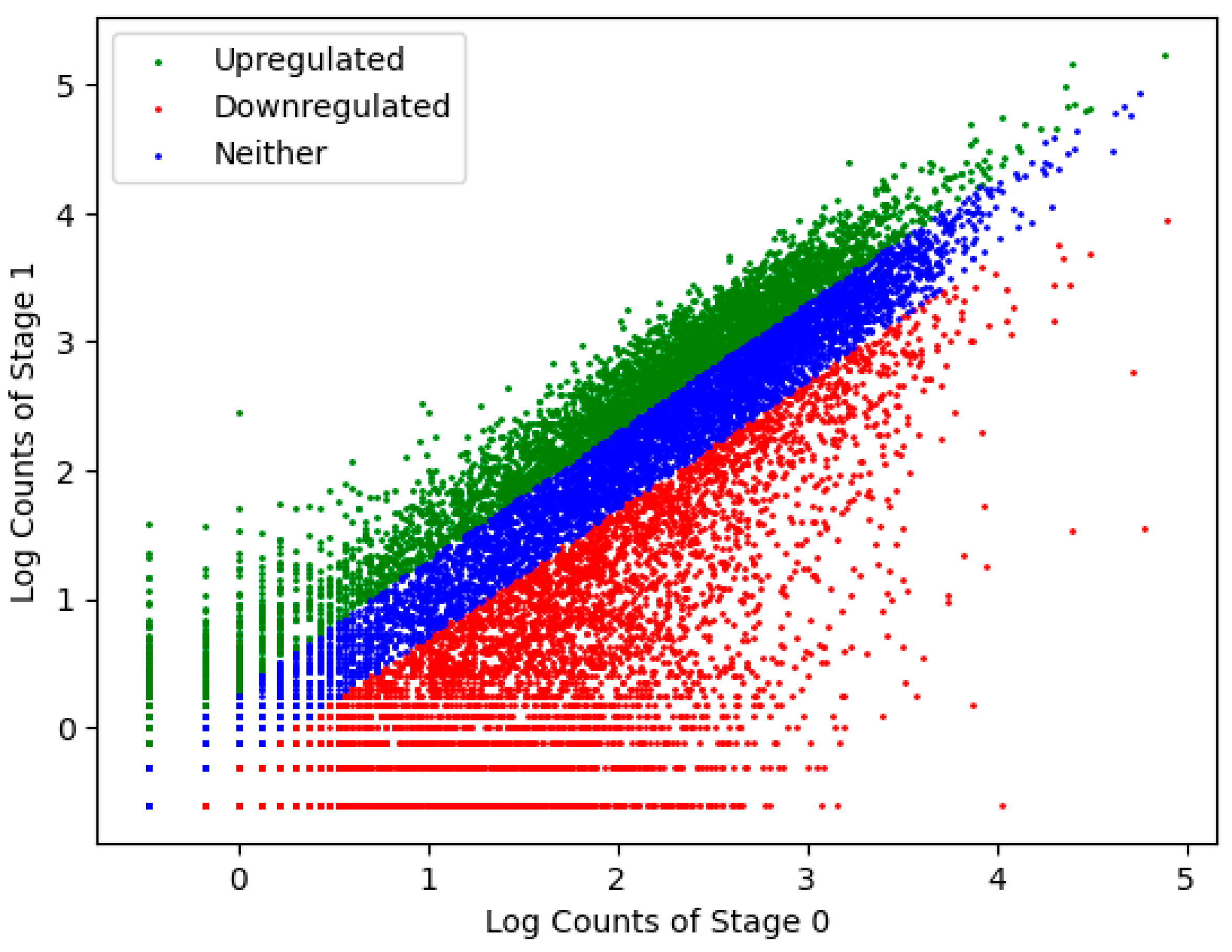
2.2. Transcriptome Construction and Analysis
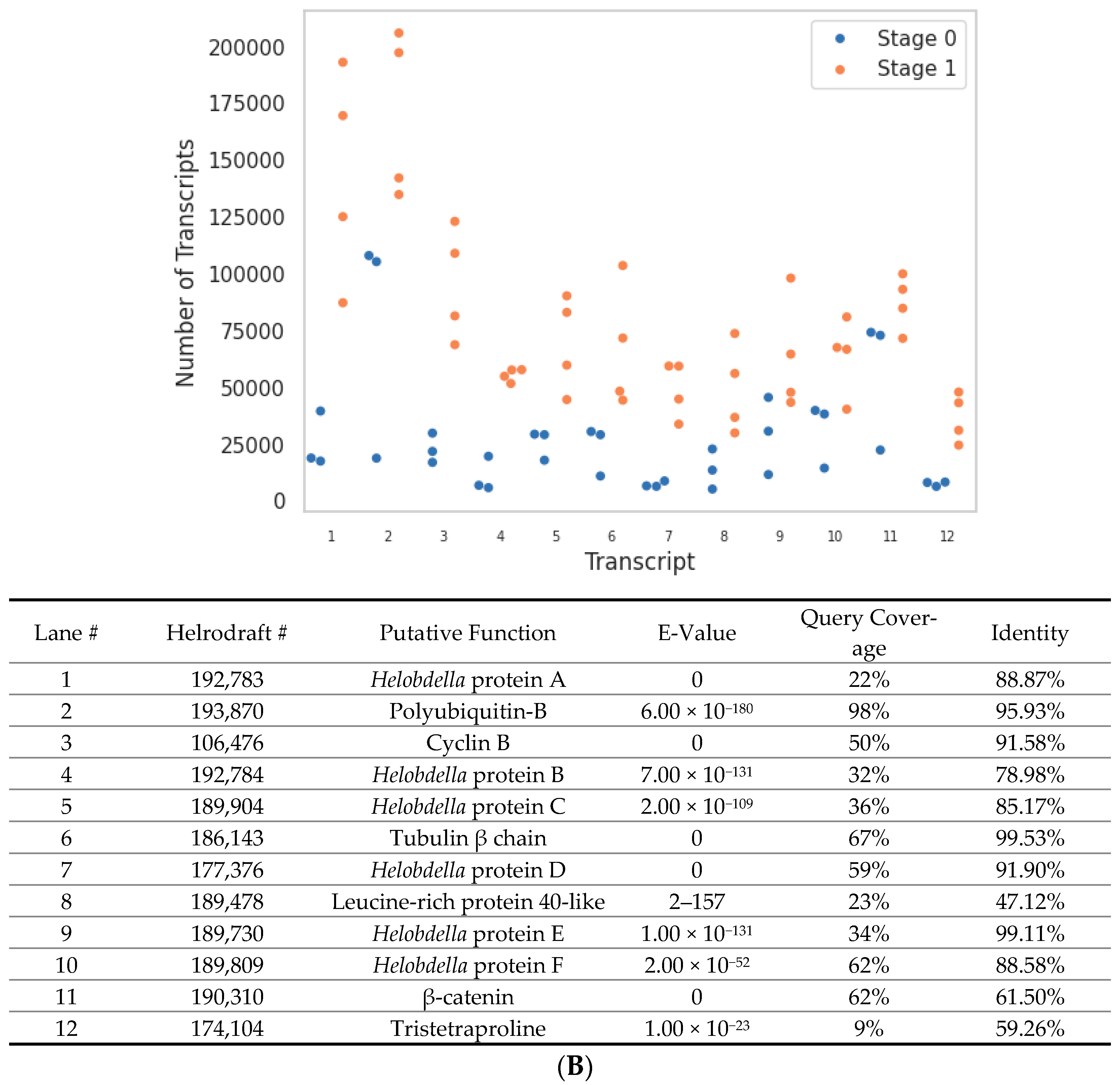
2.3. Gene Searches
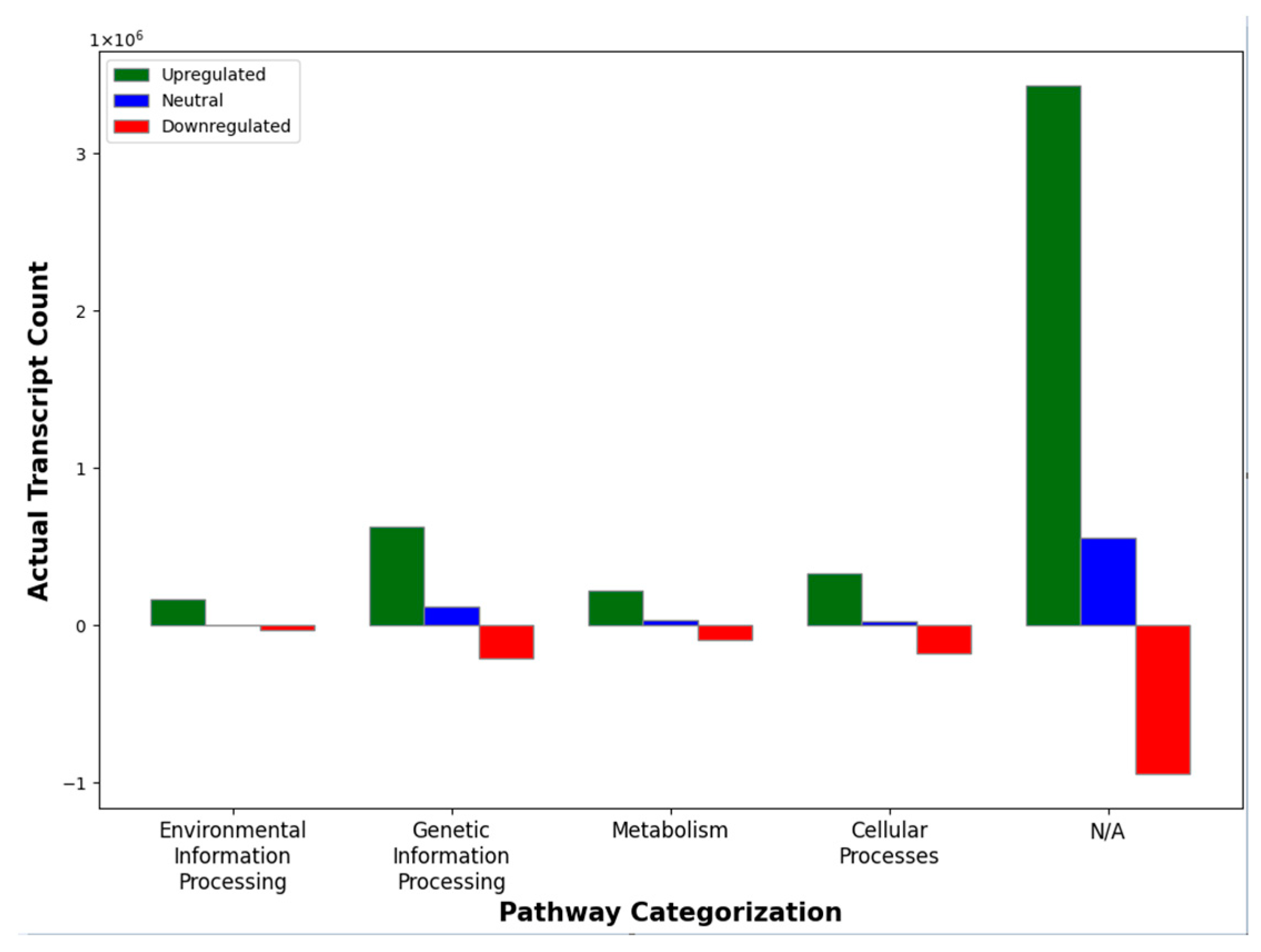
2.4. Classification Using KEGG
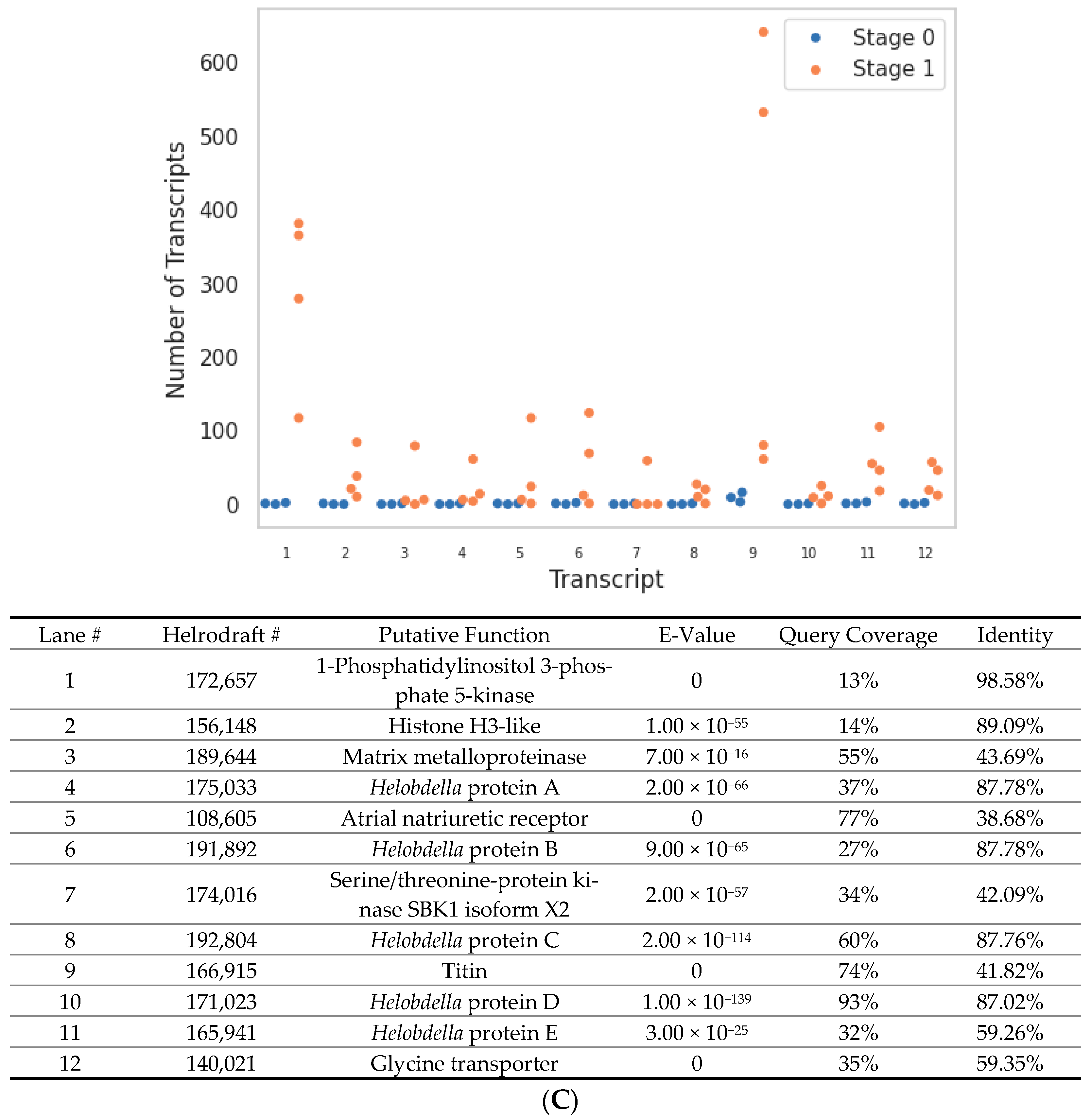
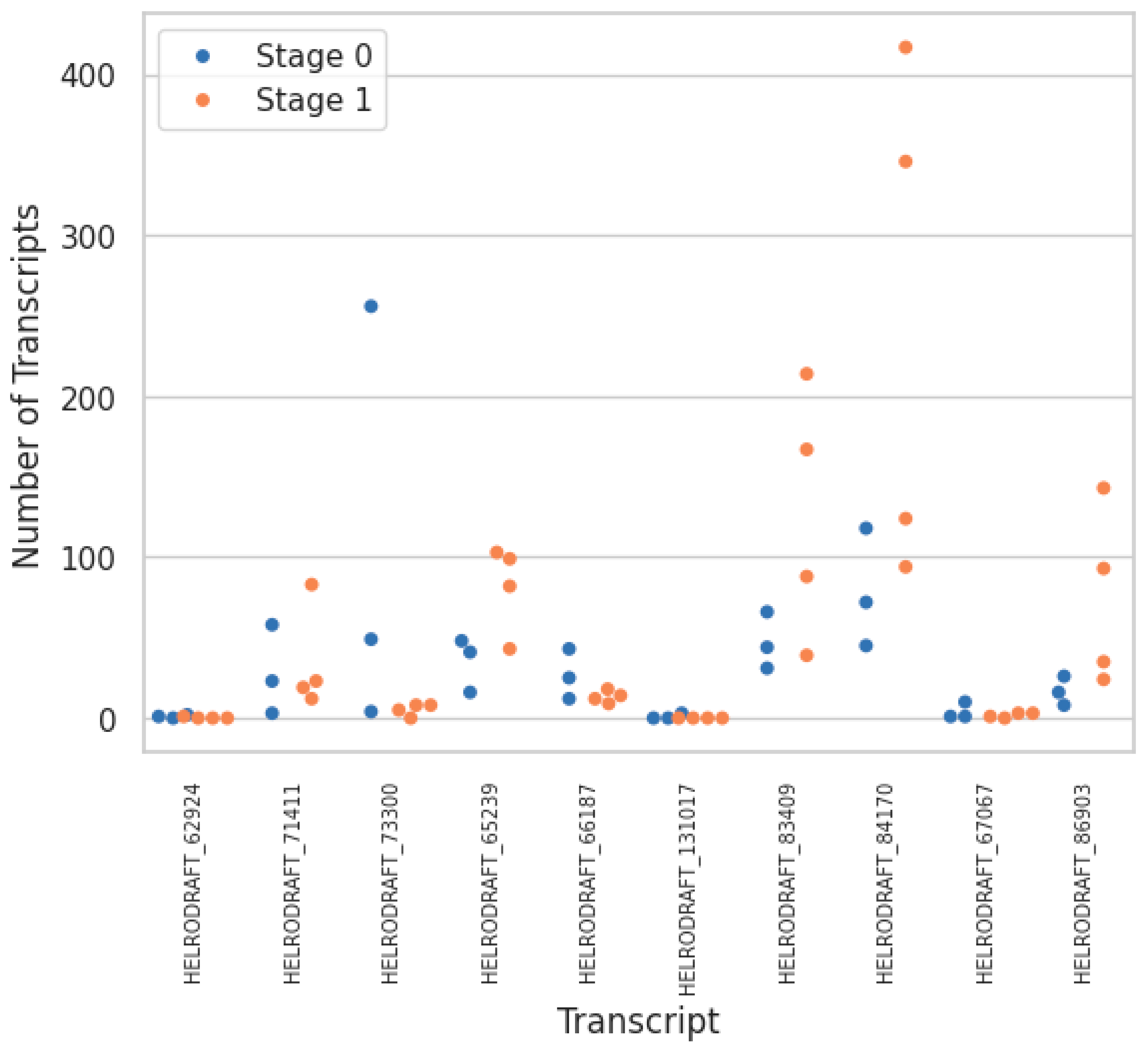
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BLAST | basic local alignment search tool |
| Hro-nos | Helobdella robusta nanos homologue |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MZT | maternal-to-zygotic transition |
| RNA | ribonucleic acid |
References
- Aguinaldo, A.M.A.; Turbeville, J.M.; Linford, L.S.; Rivera, M.C.; Garey, J.R.; Raff, R.A.; Lake, J.A. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 1997, 387, 489–493. [Google Scholar] [CrossRef]
- Ruiz-Trillo, I.; Riutort, M.; Littlewood, D.T.; Herniou, E.A.; Baguna, J. Acoel flatworms: Earliest extant bilaterian Metazoans, not members of Platyhelminthes. Science 1999, 283, 1919–1923. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, K.A.; Shain, D.H. Changes in gene expression at the precursor→ stem cell transition in leech. Stem Cells 2004, 22, 514–521. [Google Scholar] [CrossRef]
- Weisblat, D.A.; Kuo, D.H. Developmental biology of the leech Helobdella. Internat. J. Dev. Biol. 2014, 58, 429. [Google Scholar] [CrossRef] [PubMed]
- Weisblat, D.A.; Zackson, S.L.; Blair, S.S.; Young, J.D. Cell lineage analysis by intracellular injection of fluorescent tracers. Science 1980, 209, 1538–1541. [Google Scholar] [CrossRef]
- Kim, R.C.; Le, D.; Ma, K.; Heath-Heckman, E.A.C.; Whitehorn, N.; Kristan, W.B., Jr.; Weisblat, D.A. Behavioral analysis of substrate texture preference in a leech, Helobdella austinensis. J. Comp. Phys. A Neuroethol. Sens. Neural Behav. Physiol. 2019, 205, 191–202. [Google Scholar] [CrossRef]
- Saglam, N.; Melissaratos, D.S.; Shain, D.H. Biocontrol of snail-borne parasites with the glossiphoniid leech, Helobdella austinensis. Biol. Lett. 2023, 19, 20220484. [Google Scholar] [CrossRef] [PubMed]
- Astrow, S.H.; Holton, B.; Weisblat, D.A. Teloplasm formation in a leech, Helobdella triserialis, is a microtubule-dependent process. Dev. Biol. 1989, 135, 306–319. [Google Scholar] [CrossRef]
- Ren, X.; Weisblat, D.A. Asymmetrization of first cleavage by transient disassembly of one spindle pole aster in the leech Helobdella robusta. Dev. Biol. 2006, 292, 103–115. [Google Scholar] [CrossRef]
- Weisblat, D.A. Asymmetric cell divisions in the early embryo of the leech Helobdella robusta. Asymmetric Cell Div. 2007, 134, 79–95. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014, 15, 1080–1186. [Google Scholar]
- Liang, S.; Wang, B.; Pan, L.; Ye, Y.; He, M.; Han, S.; Zheng, S.; Wang, X.; Lin, Y. Comprehensive structural annotation of Pichia pastoris transcriptome and the response to various carbon sources using deep paired-end RNA sequencing. BMC Genom. 2012, 13, 738. [Google Scholar] [CrossRef] [PubMed]
- Baugh, L.R.; Hill, A.A.; Slonim, D.K.; Brown, E.L.; Hunter, C.P. Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development 2003, 130, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Tomari, Y. Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol. Cell 2016, 61, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, S.; Anders, S.; Janga, S.C.; Huber, W.; Alonso, C.R. Genome-wide analysis of mRNA decay patterns during early Drosophila development. Genome Biol. 2010, 11, R93. [Google Scholar] [CrossRef]
- Vastenhouw, N.L.; Cao, W.X.; Lipshitz, H.D. The maternal-to-zygotic transition revisited. Development 2019, 146, dev161471. [Google Scholar] [CrossRef]
- Lee, M.T.; Bonneau, A.R.; Giraldez, A.J. Zygotic genome activation during the maternal-to-zygotic transition. Ann. Rev. Cell Dev. Biol. 2014, 30, 581–613. [Google Scholar] [CrossRef]
- Laver, J.D.; Li, X.; Ray, D.; Cook, K.B.; Hahn, N.A.; Nabeel-Shah, S.; Kekis, M.; Luo, H.; Marsolais, A.J.; Fung, K.Y.; et al. Brain tumor is a sequence- specific RNA-binding protein that directs maternal mRNA clearance during the Drosophila maternal-to-zygotic transition. Genome Biol. 2015, 16, 94. [Google Scholar] [CrossRef]
- Wreden, C.; Verrotti, A.C.; Schisa, J.A.; Lieberfarb, M.E.; Strickland, S. Nanos and pumilio establish embryonic polarity in Drosophila by promoting posterior deadenylation of hunchback mRNA. Development 1997, 124, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Agee, S.J.; Lyons, D.C.; Weisblat, D.A. Maternal expression of a NANOS homolog is required for early development of the leech Helobdella robusta. Dev. Biol. 2006, 298, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pilon, M.; Weisblat, D.A. A nanos homolog in leech. Development 1997, 124, 1771–1780. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript Type | Unique Transcripts | Change in Total Transcript Count between Stage 0 → 1 |
|---|---|---|
| Downregulated (degraded) | 8295 | −1,449,456 |
| Upregulated (transcribed) | 4979 | +4,785,366 |
| Stage 0—specific | 3322 | −93,259 |
| Stage 1—specific | 760 | +1122 |
| Homologue | Helrodraft # | Query Coverage | E-Value | Identity | Stage 0 Count | Stage 1 Count |
|---|---|---|---|---|---|---|
| Par1 | 171,411 | 28% | 0 | 79.23% | 28 | 34 |
| Par3 | 192,383 | 33% | 5 × 10−53 | 31.02% | 191 | 499 |
| Par6 | 106,160 | 69% | 3 × 10−92 | 59.68% | 0 | 0 |
| SKN-1 | 130,334 | 53.0% | 2 × 10−83 | 45.45% | 7 | 0 |
| Bicoid | 148,580 | 9.00% | 1 × 10−12 | 46.55% | 0 | 0 |
| Hunchback | 116,286 | 34% | 3 × 10−47 | 54.49% | 79 | 20 |
| Caudal | 112,342 | 81% | 1 × 10−165 | 34.15% | 128 | 250 |
| Nanos | 166,185 | 85% | 1 × 10−140 | 48.11% | 519 | 1114 |
| Polarity protein | 169,490 | 42% | 4 × 10−113 | 56.15% | 80 | 583 |
| Rho1 | 167,950 | 100% | 1 × 10−126 | 87.50% | 2215 | 4302 |
| Rho4 | 161,341 | 41.0% | 2 × 10−32 | 40.0% | 1786 | 2153 |
| Wingless | 163,913 | 66% | 5 × 10−53 | 41.40% | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsaio, S.; Saglam, N.; Morrow, D.; Shain, D.H. Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis. Genes 2024, 15, 283. https://doi.org/10.3390/genes15030283
Hsaio S, Saglam N, Morrow D, Shain DH. Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis. Genes. 2024; 15(3):283. https://doi.org/10.3390/genes15030283
Chicago/Turabian StyleHsaio, Samuel, Naim Saglam, David Morrow, and Daniel H. Shain. 2024. "Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis" Genes 15, no. 3: 283. https://doi.org/10.3390/genes15030283
APA StyleHsaio, S., Saglam, N., Morrow, D., & Shain, D. H. (2024). Transcriptomic Profiling at the Maternal-to-Zygotic Transition in Leech, Helobdella austinensis. Genes, 15(3), 283. https://doi.org/10.3390/genes15030283