



Cross-Omic Transcription Factor Analysis: An Insight on Transcription Factor Accessibility and Expression Correlation



Abstract
1. Introduction
2. Background
2.1. Single-Cell Sequencing Technologies
2.2. GAGAM
3. Materials and Methods
3.1. Dataset
3.2. Aggregated Cells
3.3. TF Motifs and Motif Enrichment Analysis
3.4. TF Footprints
| Algorithm 1: Footprint score computation |
|
3.5. Correlation with Expression
4. Results
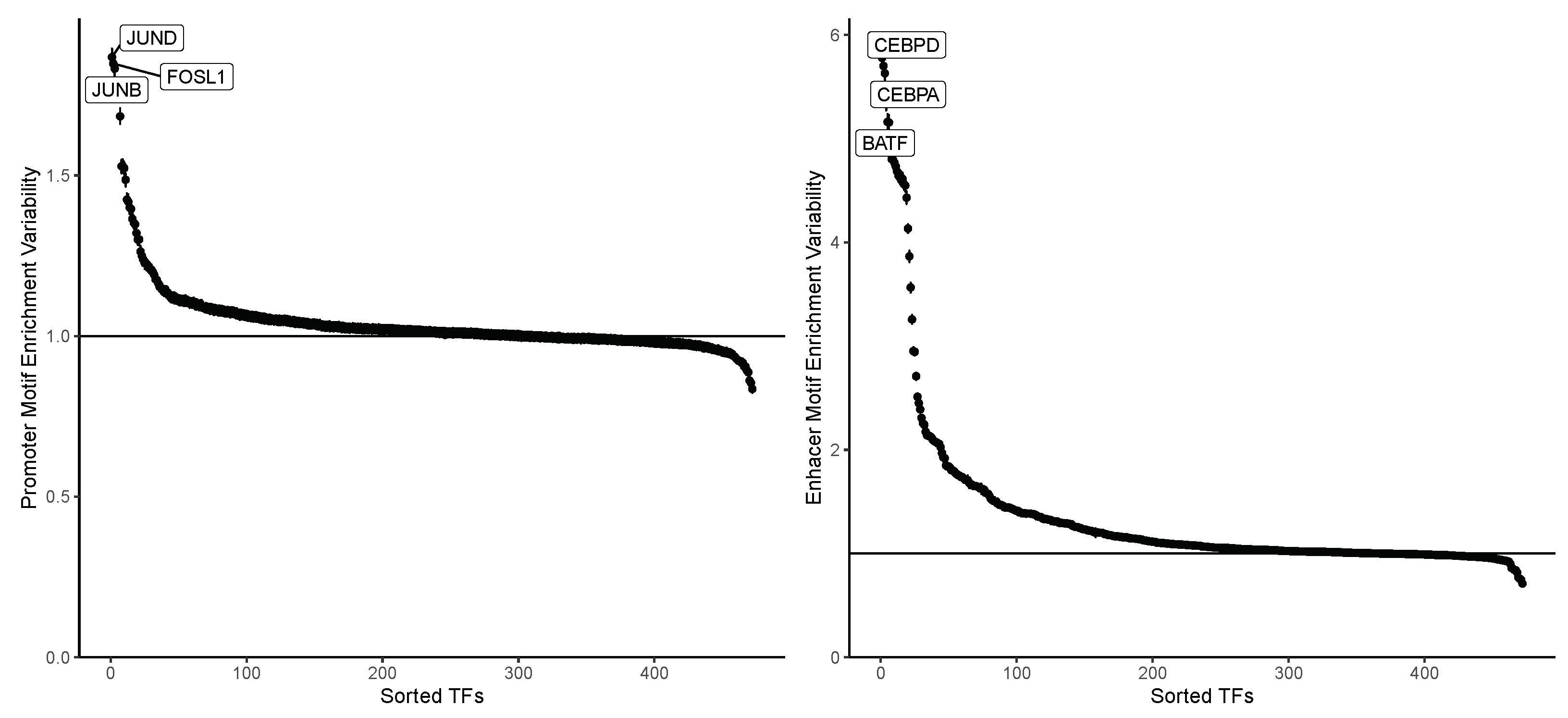
4.1. Enhancer Regions Shows More Variability in Motif Information
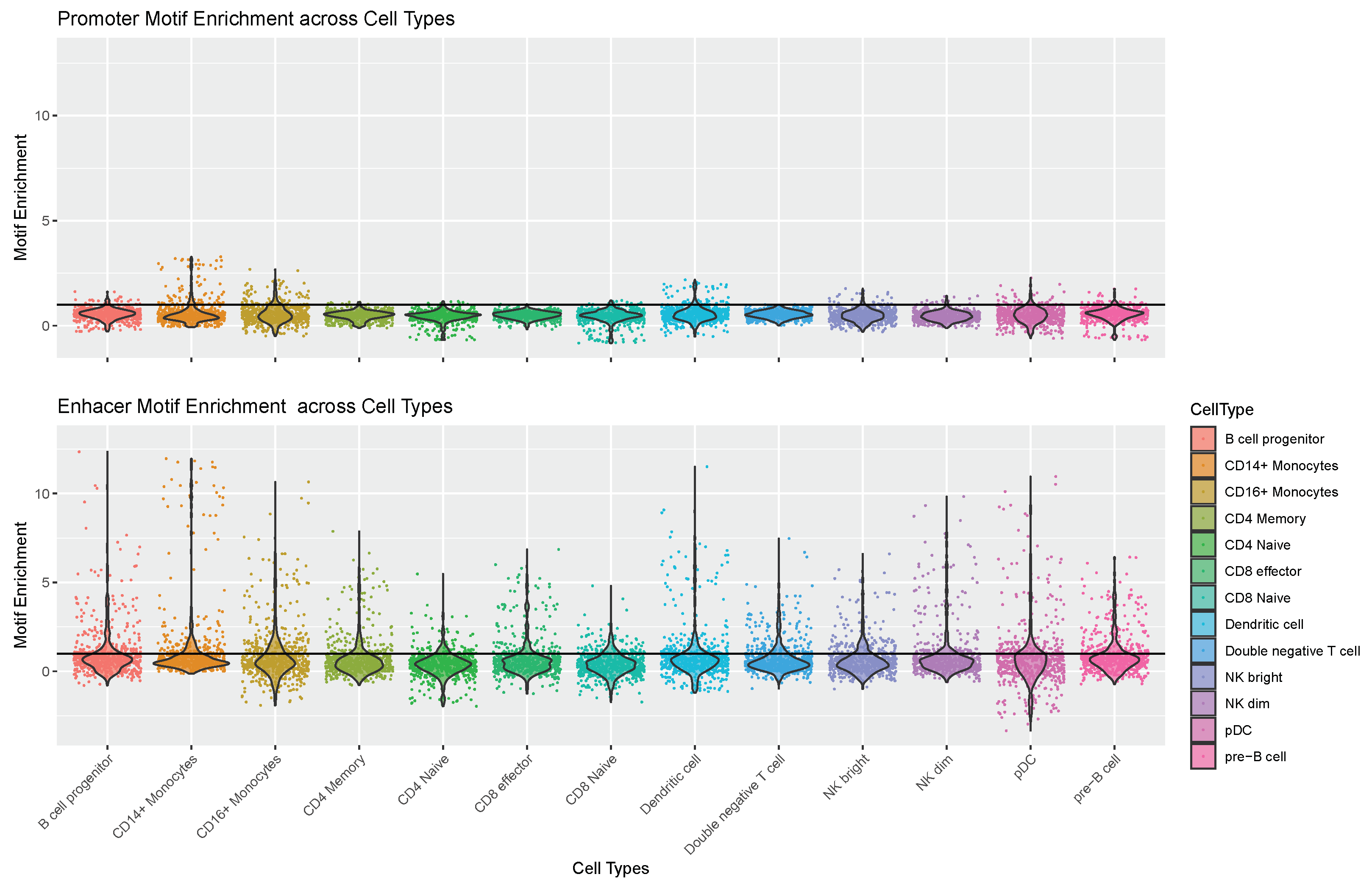
4.2. General Correlation with Expression Is Low
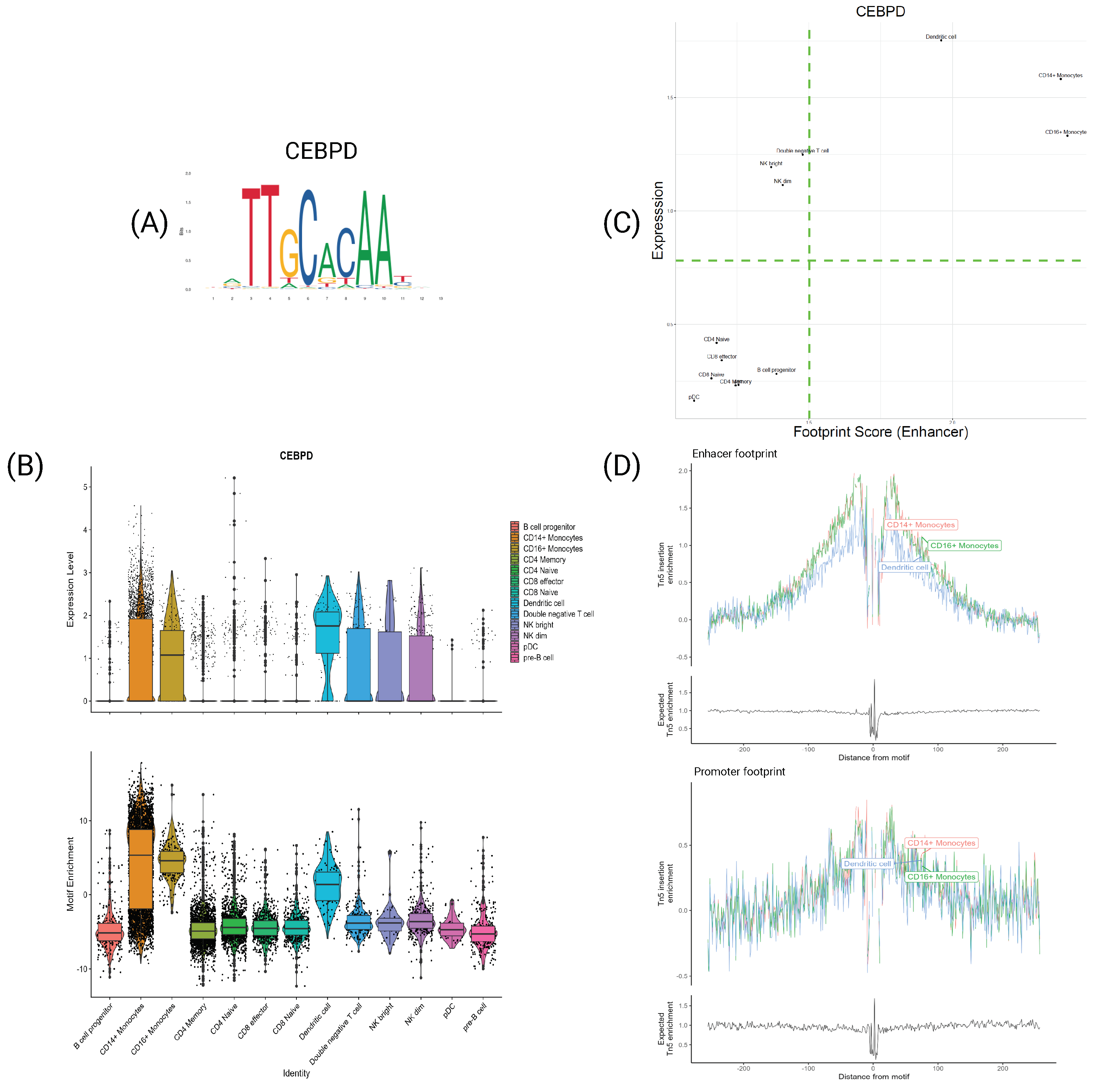
4.3. Motif Information Highlights Differences in AP-1 Subunits
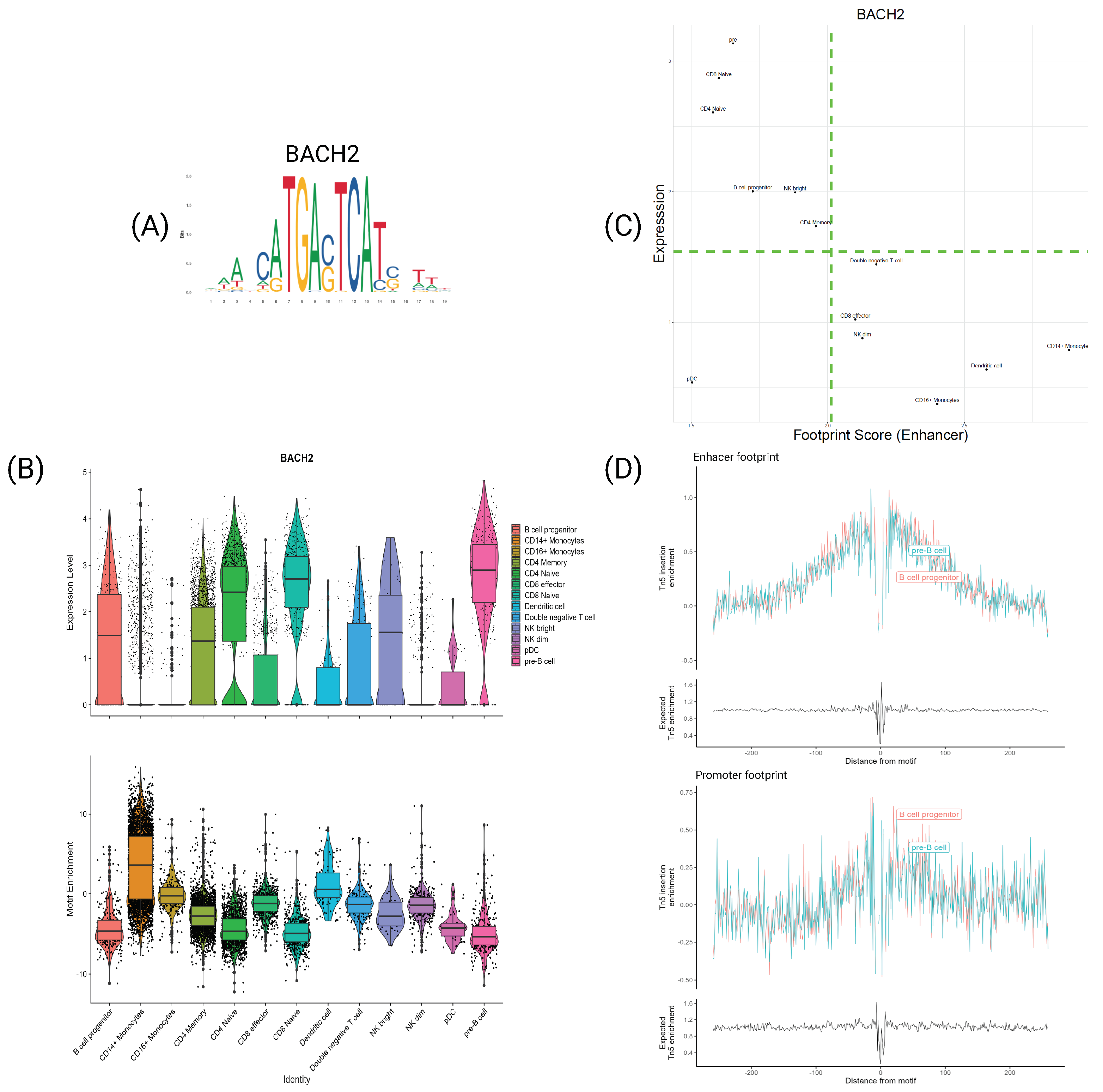
4.4. Specific TFs Shows Differences Only at the Expression Levels
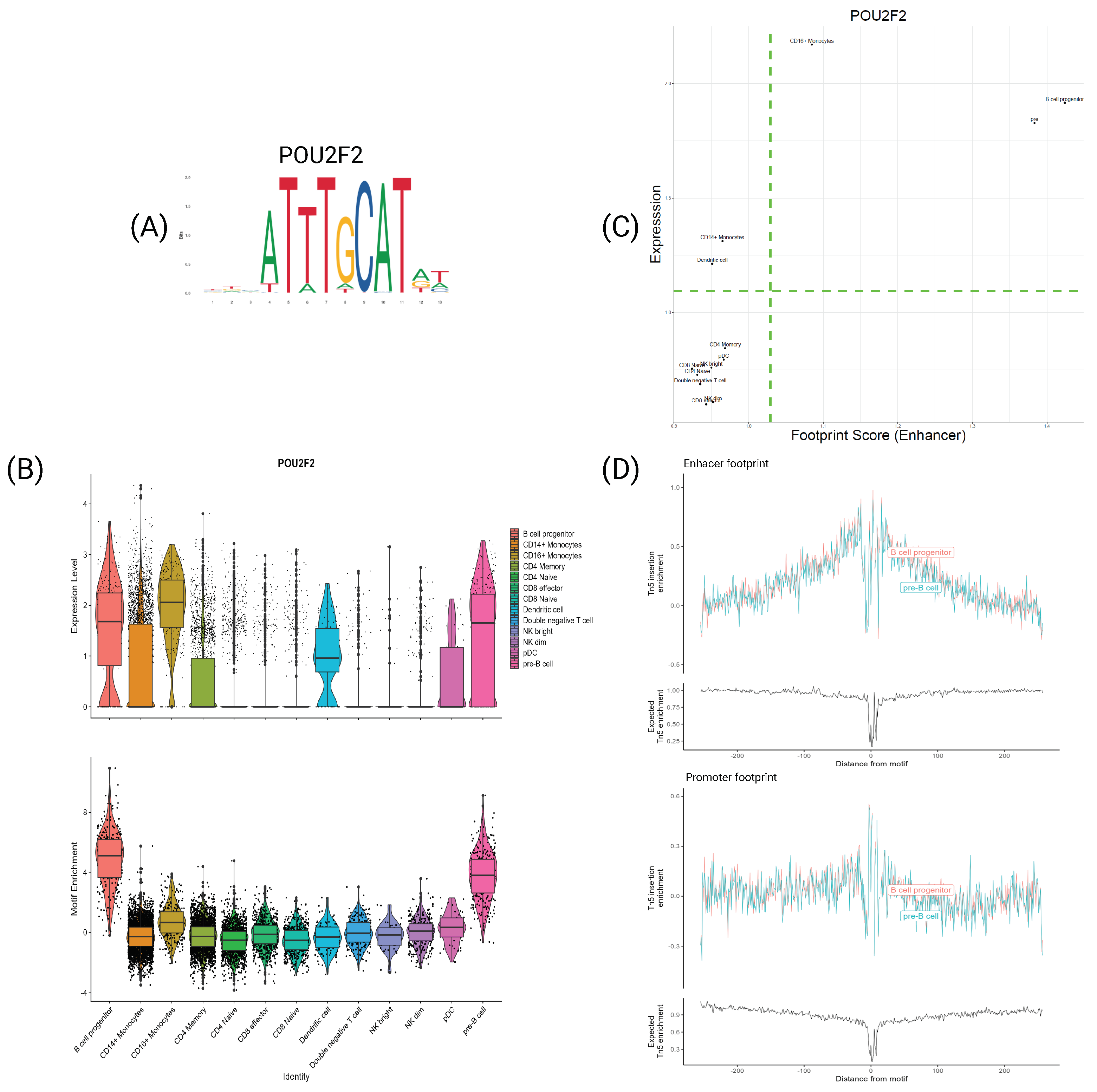
4.5. TFs Characterize Cell Types at Both Expression and Motif Information Levels
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Martini, L.; Bardini, R.; Di Carlo, S. Meta-Analysis of cortical inhibitory interneurons markers landscape and their performances in scRNA-seq studies. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 253–258. [Google Scholar] [CrossRef]
- Martini, L.; Amprimo, G.; Di Carlo, S.; Olmo, G.; Ferraris, C.; Savino, A.; Bardini, R. Neuronal Spike Shapes (NSS): A straightforward approach to investigate heterogeneity in neuronal excitability states. Comput. Biol. Med. 2024, 168, 107783. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Corces, M.R.; Lareau, C.A.; Wu, B.; Schep, A.N.; Aryee, M.J.; Majeti, R.; Chang, H.Y.; Greenleaf, W.J. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 2018, 173, 1535–1548.e16. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Lee, I. Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 2020, 18, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871. [Google Scholar] [CrossRef]
- Martini, L.; Bardini, R.; Savino, A.; Di Carlo, S. GAGAM v1.2: An Improvement on Peak Labeling and Genomic Annotated Gene Activity Matrix Construction. Genes 2023, 14, 115. [Google Scholar] [CrossRef]
- Martini, L.; Bardini, R.; Savino, A.; Di Carlo, S. GAGAM: A Genomic Annotation-Based Enrichment of scATAC-seq Data for Gene Activity Matrix. In Proceedings of the Bioinformatics and Biomedical Engineering; Rojas, I., Valenzuela, O., Rojas, F., Herrera, L.J., Ortuño, F., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 18–32. [Google Scholar]
- Martini, L.; Savino, A.; Bardini, R.; Carlo, S.D. GRAIGH: Gene Regulation accessibility integrating GeneHancer database. bioRxiv 2023. [Google Scholar] [CrossRef]
- Martini, L.; Bardini, R.; Savino, A.; Di Carlo, S. Meta-analysis of Gene Activity (MAGA) Contributions and Correlation with Gene Expression, Through GAGAM. In Proceedings of the Bioinformatics and Biomedical Engineering; Springer Nature: Cham, Switzerland, 2023; pp. 193–207. [Google Scholar]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef]
- Danese, A.; Richter, M.L.; Chaichoompu, K.; Fischer, D.S.; Theis, F.J.; Colomé-Tatché, M. EpiScanpy: Integrated single-cell epigenomic analysis. Nat. Commun. 2021, 12, 5228. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Duarte, F.M.; Chew, J.G.; Kartha, V.K.; Burkett, Z.D.; Kohlway, A.S.; Pokholok, D.; Aryee, M.J.; Steemers, F.J.; Lebofsky, R.; et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 2019, 37, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of Computational Methods for the Analysis of Single-Cell ATAC-Seq Data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- 10XGenomics. 10k Peripheral Blood Mononuclear Cells (PBMCs) from a Healthy Donor Single Cell Multiome ATAC + Gene Expression Dataset by Cell Ranger ARC 2.0.0. Available online: https://www.10xgenomics.com/datasets/10-k-human-pbm-cs-multiome-v-1-0-chromium-controller-1-standard-2-0-0 (accessed on 9 August 2021).
- Hwang, B.; Lee, J.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Riudavets-Puig, R.; Blanc-Mathieu, R.; Castro-Mondragon, J.A.; Ferenc, K.; Kumar, V.; Lemma, R.B.; Lucas, J.; Chèneby, J.; Baranasic, D.; et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2023, 52, D174–D182. [Google Scholar] [CrossRef]
- Schep, A.N.; Wu, B.; Buenrostro, J.D.; Greenleaf, W.J. chromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 2017, 14, 975–978. [Google Scholar] [CrossRef]
- Lee, B.K.; Bhinge, A.A.; Battenhouse, A.; McDaniell, R.M.; Liu, Z.; Song, L.; Ni, Y.; Birney, E.; Lieb, J.D.; Furey, T.S.; et al. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome Res. 2012, 22, 9–24. [Google Scholar] [CrossRef]
- Holwerda, S.J.B.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, M.; Jagannathan, S.; Vallabh, S.; Kartashov, A.V.; Chen, X.; Weirauch, M.T.; Barski, A. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med. 2020, 217, e20182009. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 transcription factors as regulators of immune responses in cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef]
- Tsao, H.W.; Kaminski, J.; Kurachi, M.; Barnitz, R.A.; DiIorio, M.A.; LaFleur, M.W.; Ise, W.; Kurosaki, T.; Wherry, E.J.; Haining, W.N.; et al. Batf-mediated epigenetic control of effector CD8+ T cell differentiation. Sci. Immunol. 2022, 7, eabi4919. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, M.; Barnitz, R.A.; Yosef, N.; Odorizzi, P.M.; DiIorio, M.A.; Lemieux, M.E.; Yates, K.; Godec, J.; Klatt, M.G.; Regev, A.; et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014, 15, 373–383. [Google Scholar] [CrossRef]
- Ochiai, K.; Igarashi, K. Exploring novel functions of BACH2 in the acquisition of antigen-specific antibodies. Int. Immunol. 2023, 35, 257–265. [Google Scholar] [CrossRef]
- Kaiser, F.M.P.; Janowska, I.; Menafra, R.; de Gier, M.; Korzhenevich, J.; Pico-Knijnenburg, I.; Khatri, I.; Schulz, A.; Kuijpers, T.W.; Lankester, A.C.; et al. IL-7 receptor signaling drives human B-cell progenitor differentiation and expansion. Blood 2023, 142, 1113–1130. [Google Scholar] [CrossRef]
- Itoh-Nakadai, A.; Hikota, R.; Muto, A.; Kometani, K.; Watanabe-Matsui, M.; Sato, Y.; Kobayashi, M.; Nakamura, A.; Miura, Y.; Yano, Y.; et al. The transcription repressors Bach2 and Bach1 promote B cell development by repressing the myeloid program. Nat. Immunol. 2014, 15, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Spek, C.A.; Aberson, H.L.; Butler, J.M.; de Vos, A.F.; Duitman, J. CEBPD potentiates the macrophage inflammatory response but CEBPD knock-out macrophages fail to identify CEBPD-dependent pro-inflammatory transcriptional programs. Cells 2021, 10, 2233. [Google Scholar] [CrossRef]
- Ko, C.Y.; Chang, W.C.; Wang, J.M. Biological roles of CCAAT/Enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, H.; Li, C.; Zhu, F.; Wang, M.; Xu, Y.; Wu, B. Expression and regulatory characteristics of peripheral blood immune cells in primary Sjögren’s syndrome patients using single-cell transcriptomic. iScience 2022, 25, 105509. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Contribution | Description |
|---|---|---|
| Contribution from promoter peaks , i.e., peaks overlapping promoter signatures from ENCODE cCREs annotation. They are linked to the genes by proximity to the nearest TSS of a protein-coding gene. | ||
| Contribution from exon peaks , i.e., peaks overlapping exon regions from the NCBI RefSeq Genes annotation. They are linked to the genes they are in. | ||
| Contribution from enhancer peaks , i.e., peaks overlapping enhancer signatures from ENCODE cCREs annotation. They are linked to the genes by co-accessibility scores with promoter peaks. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, L.; Bardini, R.; Savino, A.; Di Carlo, S. Cross-Omic Transcription Factor Analysis: An Insight on Transcription Factor Accessibility and Expression Correlation. Genes 2024, 15, 268. https://doi.org/10.3390/genes15030268
Martini L, Bardini R, Savino A, Di Carlo S. Cross-Omic Transcription Factor Analysis: An Insight on Transcription Factor Accessibility and Expression Correlation. Genes. 2024; 15(3):268. https://doi.org/10.3390/genes15030268
Chicago/Turabian StyleMartini, Lorenzo, Roberta Bardini, Alessandro Savino, and Stefano Di Carlo. 2024. "Cross-Omic Transcription Factor Analysis: An Insight on Transcription Factor Accessibility and Expression Correlation" Genes 15, no. 3: 268. https://doi.org/10.3390/genes15030268
APA StyleMartini, L., Bardini, R., Savino, A., & Di Carlo, S. (2024). Cross-Omic Transcription Factor Analysis: An Insight on Transcription Factor Accessibility and Expression Correlation. Genes, 15(3), 268. https://doi.org/10.3390/genes15030268