

Genome and Transcriptome Analysis of the Torreya grandis WRKY Gene Family during Seed Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Materials
2.2. Identification of WRKY Genes in T. grandis
2.3. Phylogenetic Analysis of TgWRKY and AtWRKY Gene Families
2.4. Gene Structure Analysis of TgWRKY Gene Family
2.5. RNA Extraction and Transcriptome Analysis
2.6. Weighted Gene Coexpression Network Analysis (WGCNA)
3. Results
3.1. Identification and Phylogenetic Analysis of WRKY Proteins in T. grandis
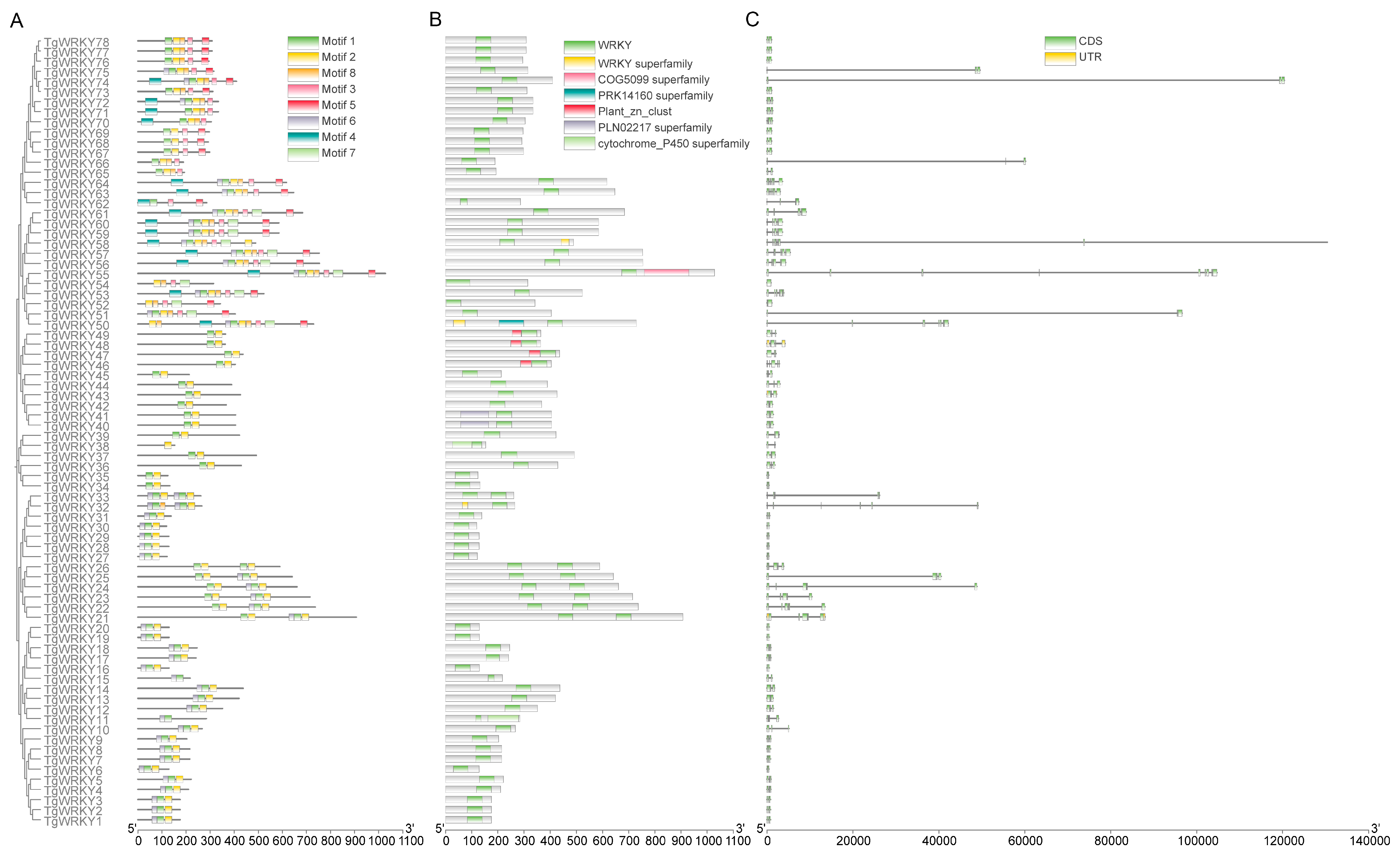
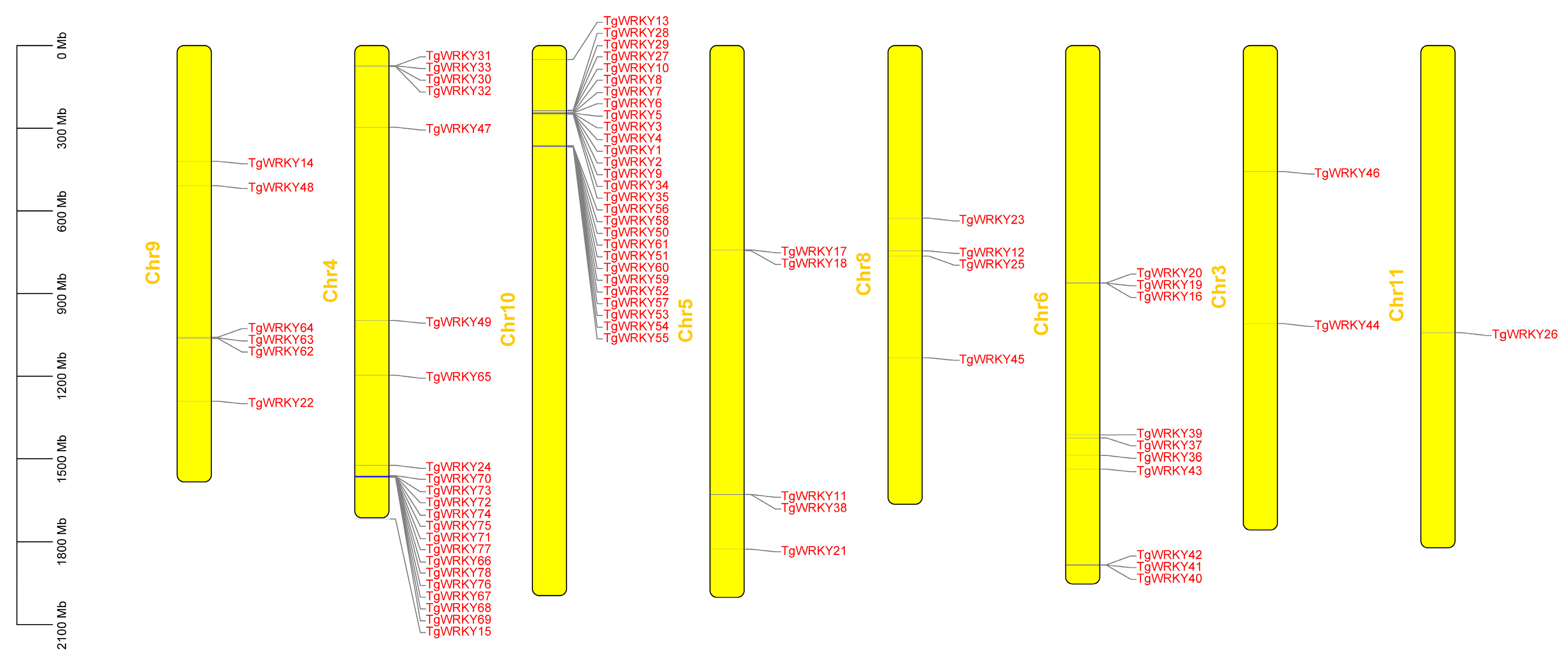
3.2. Gene Structure and Synteny Analysis of TgWRKY Gene Family
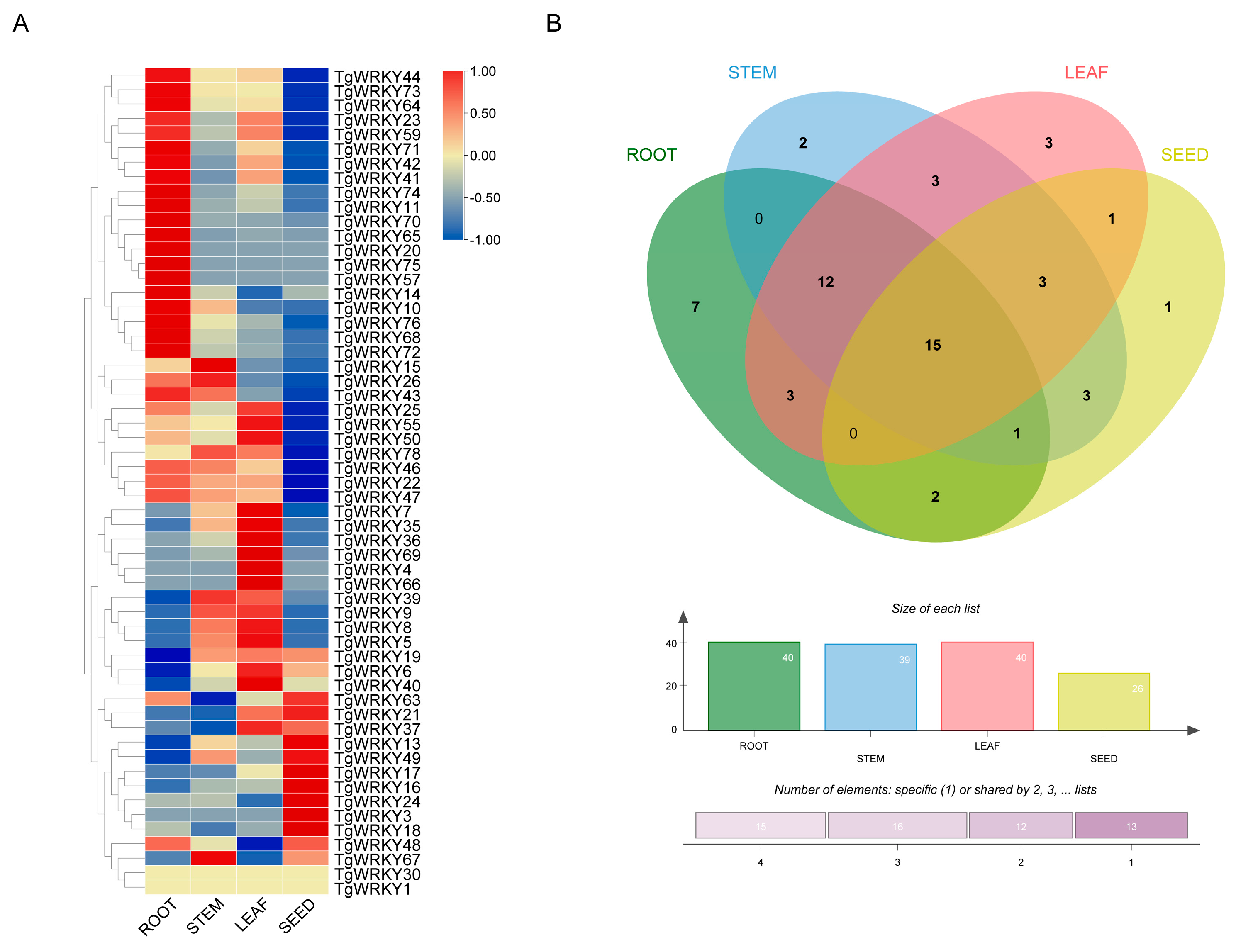
3.3. Expression Pattern Analysis of TgWRKY in Various Tissues of T. grandis
3.4. Expression Pattern Analysis of TgWRKY during the Seed Development of T. grandis
3.5. Construction of the Regulatory Network Associated with Seed Development in T. grandis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeon, B.W.; Kim, M.J.; Pandey, S.K.; Oh, E.; Seo, P.J.; Kim, J. Recent Advances in Peptide Signaling during Arabidopsis Root Development. J. Exp. Bot. 2021, 72, 2889–2902. [Google Scholar] [CrossRef] [PubMed]
- El Yahyaoui, F.; Küster, H.; Ben Amor, B.; Hohnjec, N.; Pühler, A.; Becker, A.; Gouzy, J.; Vernié, T.; Gough, C.; Niebel, A.; et al. Expression Profiling in Medicago Truncatula Identifies More than 750 Genes Differentially Expressed during Nodulation, Including Many Potential Regulators of the Symbiotic Program. Plant Physiol. 2004, 136, 3159–3176. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Nakamura, K. Characterization of a CDNA Encoding a Novel DNA-Binding Protein, SPF1, That Recognizes SP8 Sequences in the 5′ Upstream Regions of Genes Coding for Sporamin and β-Amylase from Sweet Potato. MGG Mol. Gen. Genet. 1994, 244, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Reddy, M.P.; Chikara, J. WRKY: Its Structure, Evolutionary Relationship, DNA-Binding Selectivity, Role in Stress Tolerance and Development of Plants. Mol. Biol. Rep. 2011, 38, 3883–3896. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY Superfamily of Plant Transcription Factors. Trends Plant Sci. 2000, 5, 1360–1385. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmüller, R. WRKY Transcription Factors: Jack of Many Trades in Plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, Z.L.; Zou, X.; Huang, J.; Ruas, P.; Thompson, D.; Shen, Q.J. Annotations and Functional Analyses of the Rice WRKY Gene Superfamily Reveal Positive and Negative Regulators of Abscisic Acid Signaling in Aleurone Cells. Plant Physiol. 2005, 137, 176–189. [Google Scholar] [CrossRef]
- Ülker, B.; Somssich, I.E. WRKY Transcription Factors: From DNA Binding towards Biological Function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef]
- Bencke-Malato, M.; Cabreira, C.; Wiebke-Strohm, B.; Bücker-Neto, L.; Mancini, E.; Osorio, M.B.; Homrich, M.S.; Turchetto-Zolet, A.C.; De Carvalho, M.C.C.G.; Stolf, R.; et al. Genome-Wide Annotation of the Soybean WRKY Family and Functional Characterization of Genes Involved in Response to Phakopsora pachyrhizi Infection. BMC Plant Biol. 2014, 14, 236. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, L.; Zhang, N.; Zhang, L.; Liu, Y.; Wang, Y.; Li, M.; Lin, Y.; Zhang, Y.; Zhang, Y.; et al. Importance of FaWRKY71 in Strawberry (Fragaria × Ananassa) Fruit Ripening. Int. J. Mol. Sci. 2022, 23, 12483. [Google Scholar] [CrossRef]
- Bi, C.; Xu, Y.; Ye, Q.; Yin, T.; Ye, N. Genome-Wide Identification and Characterization of WRKY Gene Family in Salix suchowensis. PeerJ 2016, 4, e2437. [Google Scholar] [CrossRef]
- Chen, D.; Lu, X.; Wu, X.; Ying, X.; Long, W.; Su, H.; Liu, H.; Lin, X.; Xu, C.; Cai, Q. Transcriptome Analysis of Axillary Bud Differentiation in a New Dual-Axillary Bud Genotype of Sugarcane. Genet. Resour. Crop Evol. 2020, 67, 685–701. [Google Scholar] [CrossRef]
- Chen, C.; Chen, X.; Han, J.; Lu, W.; Ren, Z. Genome-Wide Analysis of the WRKY Gene Family in the Cucumber Genome and Transcriptome-Wide Identification of WRKY Transcription Factors That Respond to Biotic and Abiotic Stresses. BMC Plant Biol. 2020, 20, 443. [Google Scholar] [CrossRef]
- Rosado, D.; Ackermann, A.; Spassibojko, O.; Rossi, M.; Pedmale, U.V. WRKY Transcription Factors and Ethylene Signaling Modify Root Growth during the Shade-Avoidance Response. Plant Physiol. 2022, 188, 1294–1311. [Google Scholar] [CrossRef]
- Li, W.; Tian, Z.; Yu, D. WRKY13 Acts in Stem Development in Arabidopsis thaliana. Plant Sci. 2015, 236, 205–213. [Google Scholar] [CrossRef]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY Transcription Factors and Plant Defense Responses: Latest Discoveries and Future Prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Javed, T.; Gao, S.J. WRKY Transcription Factors in Plant Defense. Trends Genet. 2023, 39, 787–801. [Google Scholar] [CrossRef]
- Zhou, S.; Zheng, W.J.; Liu, B.H.; Zheng, J.C.; Dong, F.S.; Liu, Z.F.; Wen, Z.Y.; Yang, F.; Wang, H.B.; Xu, Z.S.; et al. Characterizing the Role of TaWRKY13 in Salt Tolerance. Int. J. Mol. Sci. 2019, 20, 5712. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Lethin, J.; Blomberg, R.; Mousavi, H.; Aronsson, H. In Silico Based Screening of WRKY Genes for Identifying Functional Genes Regulated by WRKY under Salt Stress. Comput. Biol. Chem. 2019, 83, 107131. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Kang, K.; Shim, Y.; Yoo, S.C.; Paek, N.C. Inactivating Transcription Factor OsWRKY5 Enhances Drought Tolerance through Abscisic Acid Signaling Pathways. Plant Physiol. 2022, 188, 1900–1916. [Google Scholar] [CrossRef]
- Wu, H.; Ni, Z.; Yao, Y.; Guo, G.; Sun, Q. Cloning and Expression Profiles of 15 Genes Encoding WRKY Transcription Factor in Wheat (Triticum aestivem L.). Prog. Nat. Sci. 2008, 18, 697–705. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY Transcription Factors in Plant Responses to Stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A Comparative Platform for Green Plant Genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Yan, J.; Zeng, H.; Chen, W.; Zheng, S.; Luo, J.; Jiang, H.; Yang, B.; Farag, M.A.; Lou, H.; Song, L.; et al. Effects of Tree Age on Flavonoids and Antioxidant Activity in Torreya grandis Nuts via Integrated Metabolome and Transcriptome Analyses. Food Front. 2023, 4, 358–367. [Google Scholar] [CrossRef]
- Lou, H.; Yang, Y.; Zheng, S.; Ma, Z.; Chen, W.; Yu, C.; Song, L.; Wu, J. Identification of Key Genes Contributing to Amino Acid Biosynthesis in Torreya grandis Using Transcriptome and Metabolome Analysis. Food Chem. 2022, 379, 132078. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Song, L.; Li, X.; Zi, H.; Chen, W.; Gao, Y.; Zheng, S.; Fei, Z.; Sun, X.; Wu, J. The Torreya grandis Genome Illuminates the Origin and Evolution of Gymnosperm-Specific Sciadonic Acid Biosynthesis. Nat. Commun. 2023, 14, 1315. [Google Scholar] [CrossRef]
- Zhang, Z.; Tao, L.; Gao, L.; Gao, Y.; Suo, J.; Yu, W.; Hu, Y.; Wei, C.; Farag, M.A.; Wu, J.; et al. Transcription Factors TgbHLH95 and TgbZIP44 Cotarget Terpene Biosynthesis Gene TgGPPS in Torreya Grandis Nuts. Plant Physiol. 2023, 193, 1161–1176. [Google Scholar] [CrossRef]
- Suo, J.; Ma, Z.; Zhao, B.; Ma, S.; Zhang, Z.; Hu, Y.; Yang, B.; Yu, W.; Wu, J.; Song, L. Metabolomics Reveal Changes in Flavor Quality and Bioactive Components in Post-Ripening Torreya grandis Nuts and the Underlying Mechanism. Food Chem. 2023, 406, 134987. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An Online Visualization and Management Tool for Customized and Annotated Phylogenetic Trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the Functional Annotation of Proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “One for All, All for One” Bioinformatics Platform for Biological Big-Data Mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shi, H.; Wang, H.; Wang, M.; Li, X. Antioxidant Activity and Chemical Composition of Torreya grandis Cv. Merrillii Seed. Nat. Prod. Commun. 2009, 4, 1565–1570. [Google Scholar] [PubMed]
- Kong, L.; Guo, H.; Sun, M. Signal Transduction during Wheat Grain Development. Planta 2015, 241, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Dante, R.A.; Larkins, B.A.; Sabelli, P.A. Cell Cycle Control and Seed Development. Front. Plant Sci. 2014, 5, 93. [Google Scholar] [CrossRef]
- Kozaki, A.; Aoyanagi, T. Molecular Aspects of Seed Development Controlled by Gibberellins and Abscisic Acids. Int. J. Mol. Sci. 2022, 23, 1876. [Google Scholar] [CrossRef] [PubMed]
- Coello, P.; Martínez-Barajas, E. Changes in Nutrient Distribution Are Part of the Mechanism That Promotes Seed Development under Severe Nutrient Restriction. Plant Physiol. Biochem. 2016, 99, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Attuluri, V.P.S.; Robert, H.S. Transcriptional Control of Arabidopsis Seed Development. Planta 2022, 255, 90. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, N.; Yoshioka, H. Post-Translational Regulation of WRKY Transcription Factors in Plant Immunity. Curr. Opin. Plant Biol. 2012, 15, 431–437. [Google Scholar] [PubMed]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY Family Gene, and HAIKU2 (IKU2), a Leucine-Rich Repeat (LRR) KINASE Gene, Are Regulators of Seed Size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nam, K.H. Regulation of Brassinosteroid Signaling by a GSK3/SHAGGY-like Kinase. Science 2002, 295, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Chen, Y.; Liu, Y.; Wu, Y.; Ren, S.; Li, L. Identification, Evolution and Expression Analysis of WRKY Gene Family in Eucommia ulmoides. Genomics 2021, 113, 3294–3309. [Google Scholar] [CrossRef]
- Wu, W.; Zhu, S.; Xu, L.; Zhu, L.; Wang, D.; Liu, Y.; Liu, S.; Hao, Z.; Lu, Y.; Yang, L.; et al. Genome-Wide Identification of the Liriodendron Chinense WRKY Gene Family and Its Diverse Roles in Response to Multiple Abiotic Stress. BMC Plant Biol. 2022, 22, 25. [Google Scholar] [CrossRef]
- Song, H.; Guo, Z.; Duan, Z.; Li, M.; Zhang, J. WRKY Transcription Factors in Arachis hypogaea and Its Donors: From Identification to Function Prediction. Plant Physiol. Biochem. 2023, 204, 108131. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome- Wide Identification and Expression Analysis of the WRKY Gene Family in Cassava. Front. Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef]
- Song, X.; Hou, X.; Zeng, Y.; Jia, D.; Li, Q.; Gu, Y.; Miao, H. Genome-Wide Identification and Comprehensive Analysis of WRKY Transcription Factor Family in Safflower during Drought Stress. Sci. Rep. 2023, 13, 16955. [Google Scholar]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY Transcription Factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Ramamoorthy, R.; Jiang, S.Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A Comprehensive Transcriptional Profiling of the WRKY Gene Family in Rice under Various Abiotic and Phytohormone Treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef]
- Innan, H.; Kim, Y. Pattern of Polymorphism after Strong Artificial Selection in a Domestication Event. Proc. Natl. Acad. Sci. USA 2004, 101, 10667–10672. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The Role of WRKY Transcription Factors in Plant Immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, Y.; Nie, L.; Jin, X.; Liao, W.; Zhao, S.; Fu, C.; Yu, L. Transcriptome-Wide Identification and Screening of WRKY Factors Involved in the Regulation of Taxol Biosynthesis in Taxus chinensis. Sci. Rep. 2018, 8, 55197. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Guo, D.; Yang, Z.P.; Tang, X.; Peng, S.Q. Genome-Wide Identification and Characterization of WRKY Gene Family in Hevea brasiliensis. Genomics 2014, 104, 14–23. [Google Scholar] [CrossRef]
- Shu, Y.; Liu, Y.; Zhang, J.; Song, L.; Guo, C. Genome-Wide Analysis of the AP2/ERF Superfamily Genes and Their Responses to Abiotic Stress in Medicago truncatula. Front. Plant Sci. 2016, 6, 1247. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Kracher, B.; Somssich, I.E. Induced Genome-Wide Binding of Three Arabidopsis WRKY Transcription Factors during Early MAMP-Triggered Immunity. Plant Cell 2017, 29, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Yu, D. Arabidopsis WRKY Transcription Factors WRKY12 and WRKY13 Oppositely Regulate Flowering under Short-Day Conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.; Gao, N.; Luo, J.; Shi, W. Genome and Transcriptome Analysis of the Torreya grandis WRKY Gene Family during Seed Development. Genes 2024, 15, 267. https://doi.org/10.3390/genes15030267
Zhu R, Gao N, Luo J, Shi W. Genome and Transcriptome Analysis of the Torreya grandis WRKY Gene Family during Seed Development. Genes. 2024; 15(3):267. https://doi.org/10.3390/genes15030267
Chicago/Turabian StyleZhu, Ruiqian, Ning Gao, Jiali Luo, and Wenhui Shi. 2024. "Genome and Transcriptome Analysis of the Torreya grandis WRKY Gene Family during Seed Development" Genes 15, no. 3: 267. https://doi.org/10.3390/genes15030267
APA StyleZhu, R., Gao, N., Luo, J., & Shi, W. (2024). Genome and Transcriptome Analysis of the Torreya grandis WRKY Gene Family during Seed Development. Genes, 15(3), 267. https://doi.org/10.3390/genes15030267