
Frameshift Variant in AMPD2 in Cirneco dell’Etna Dogs with Retinopathy and Tremors

 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Sample Collection and Phenotype Assessment
2.3. Mapping of the Causative Variant
2.3.1. Homozygosity Mapping
2.3.2. Whole-Genome Sequencing
2.3.3. SNV and Short In-Del Discovery
2.3.4. Structural Variant and Mobile Element Discovery
2.4. Genotyping
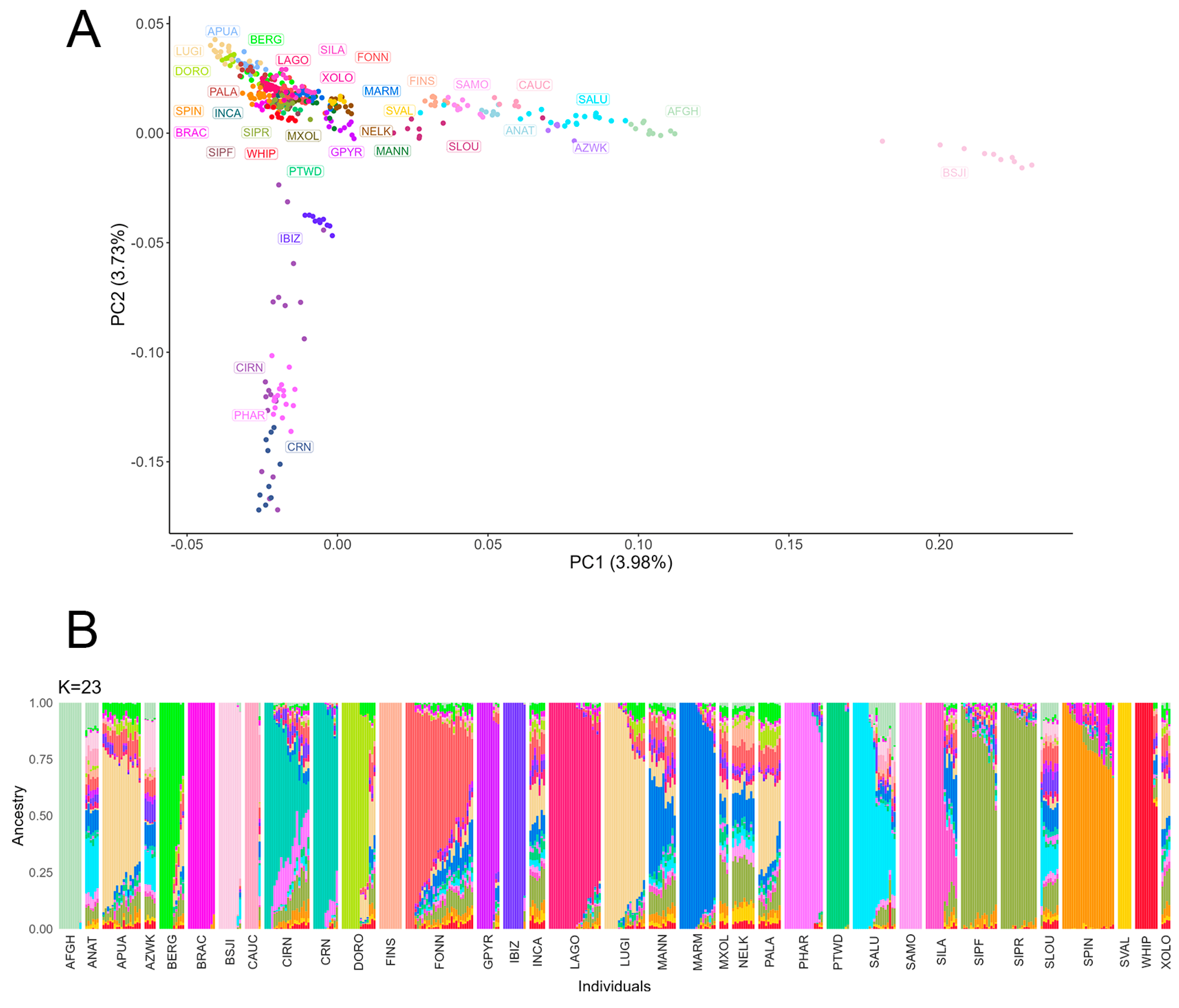
2.5. Population Structure
3. Results
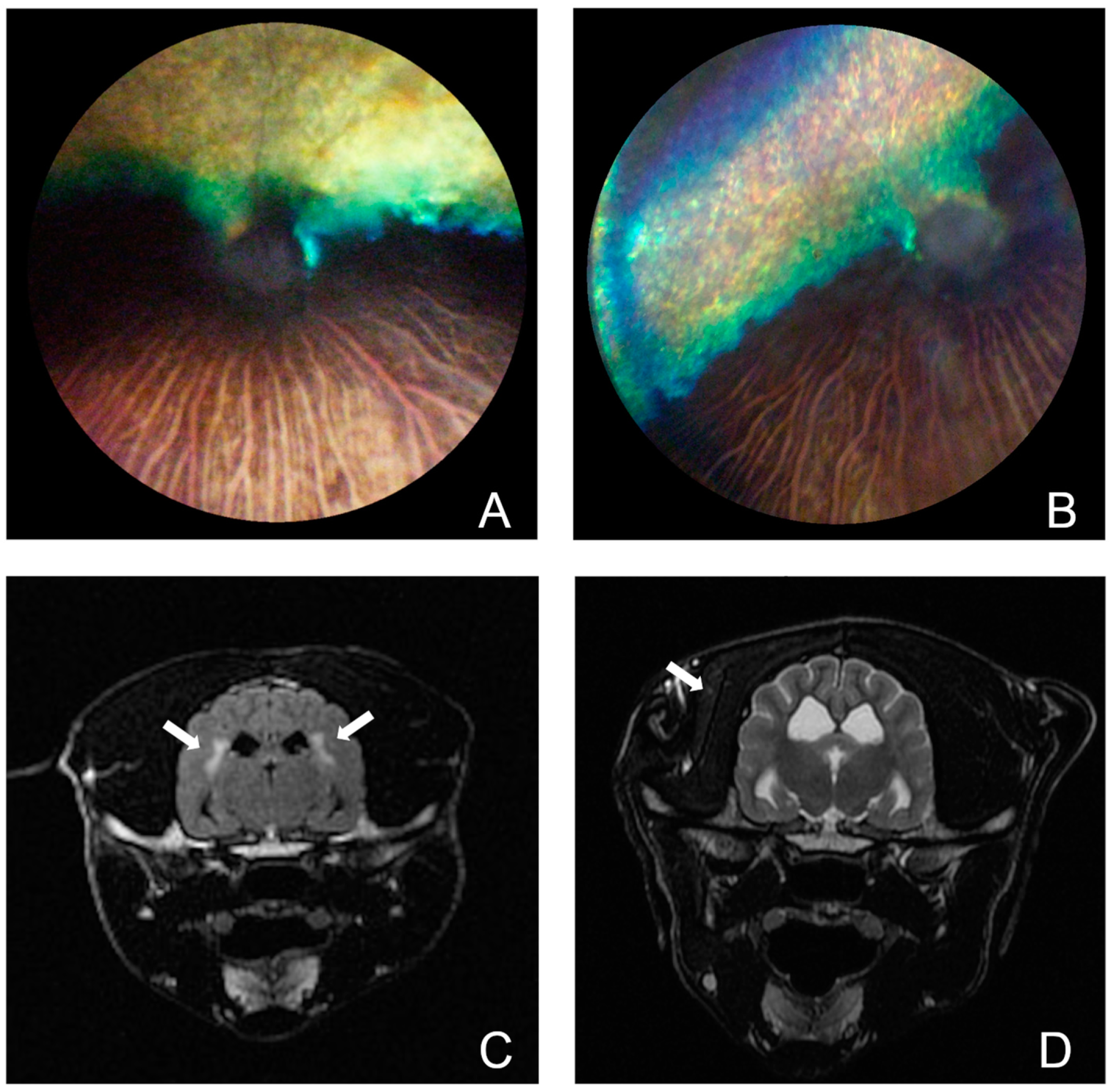
3.1. Phenotype Characterization
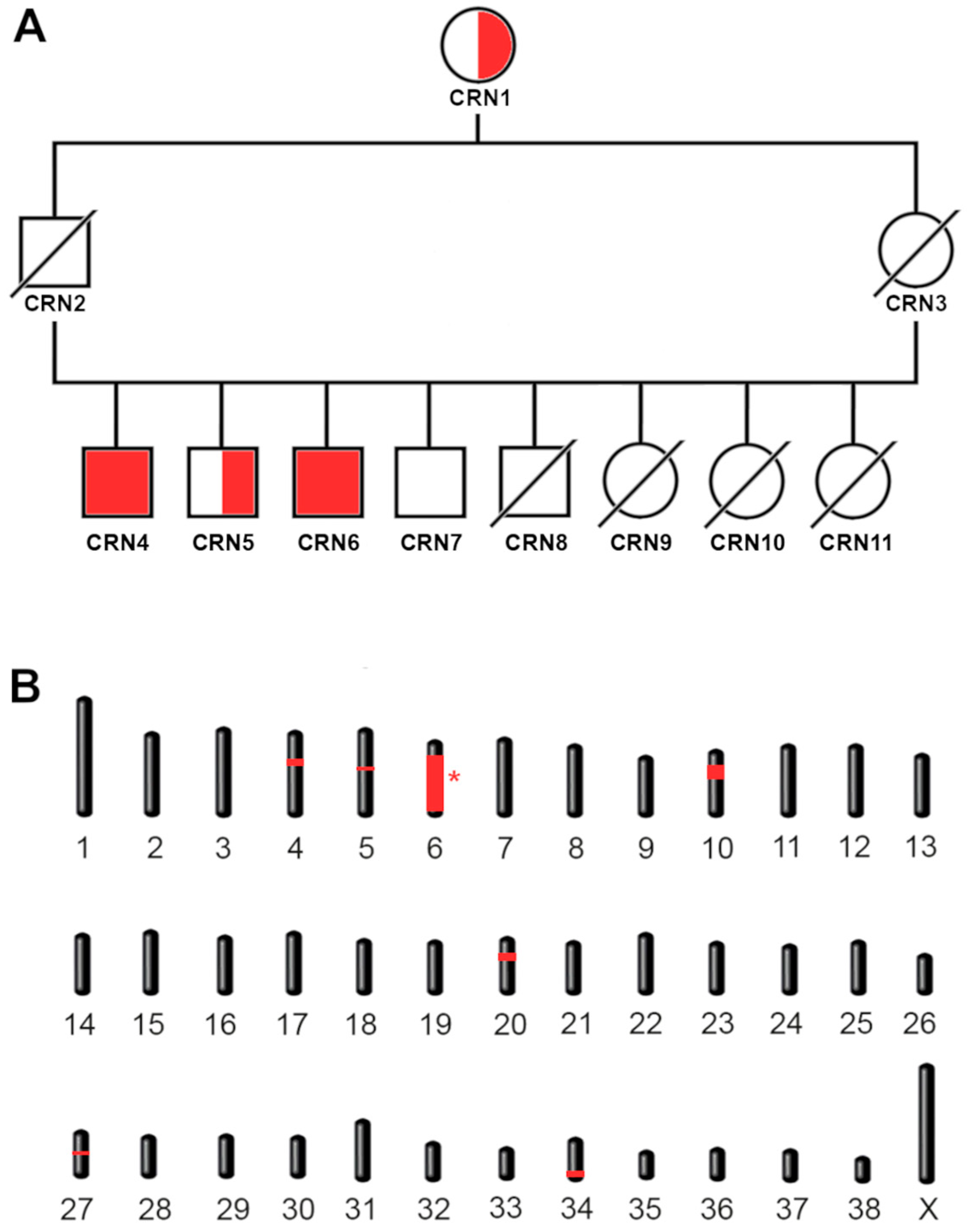
3.2. Family Tree and Inheritance Mechanism
3.3. Mapping of the Candidate Regions
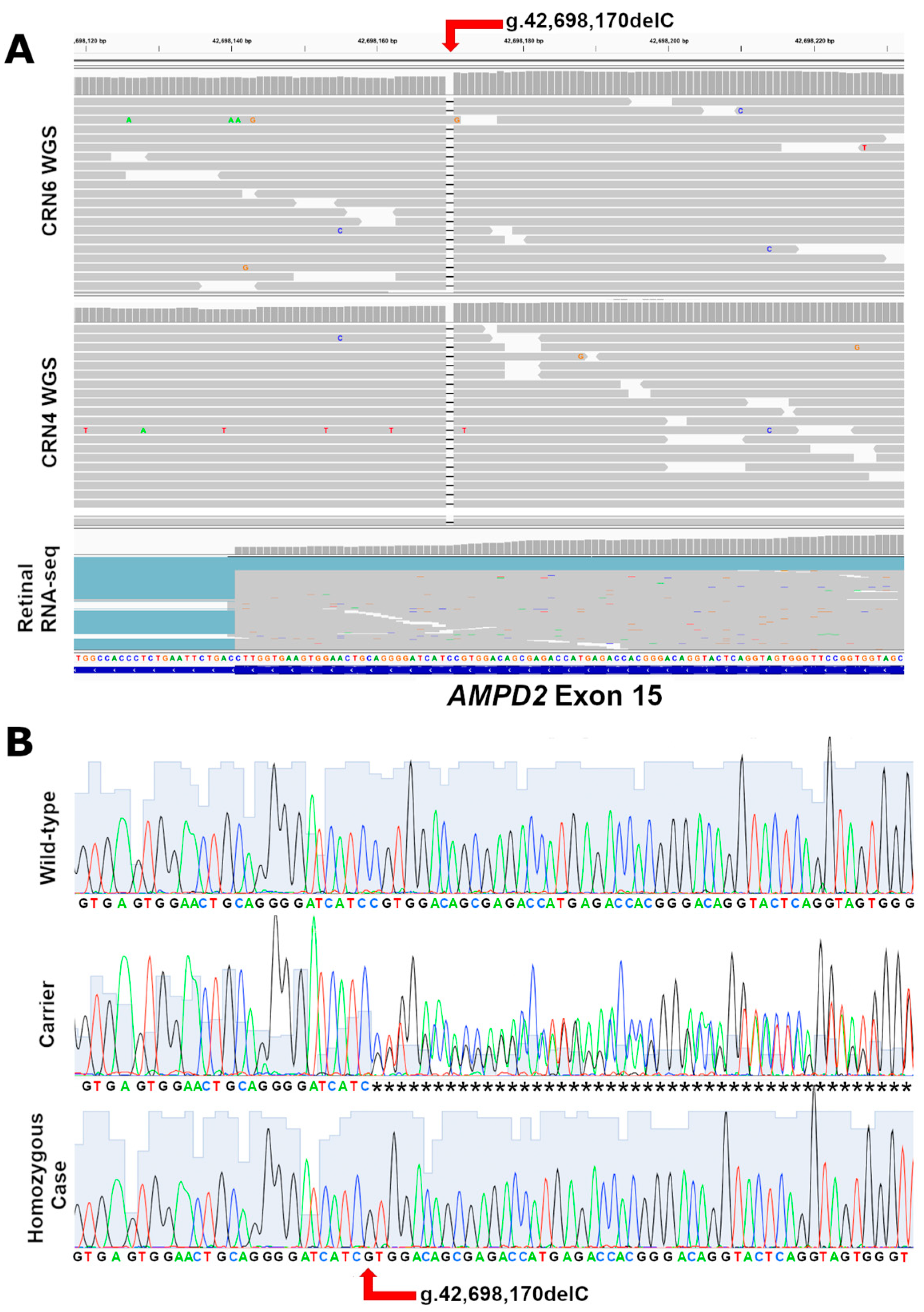
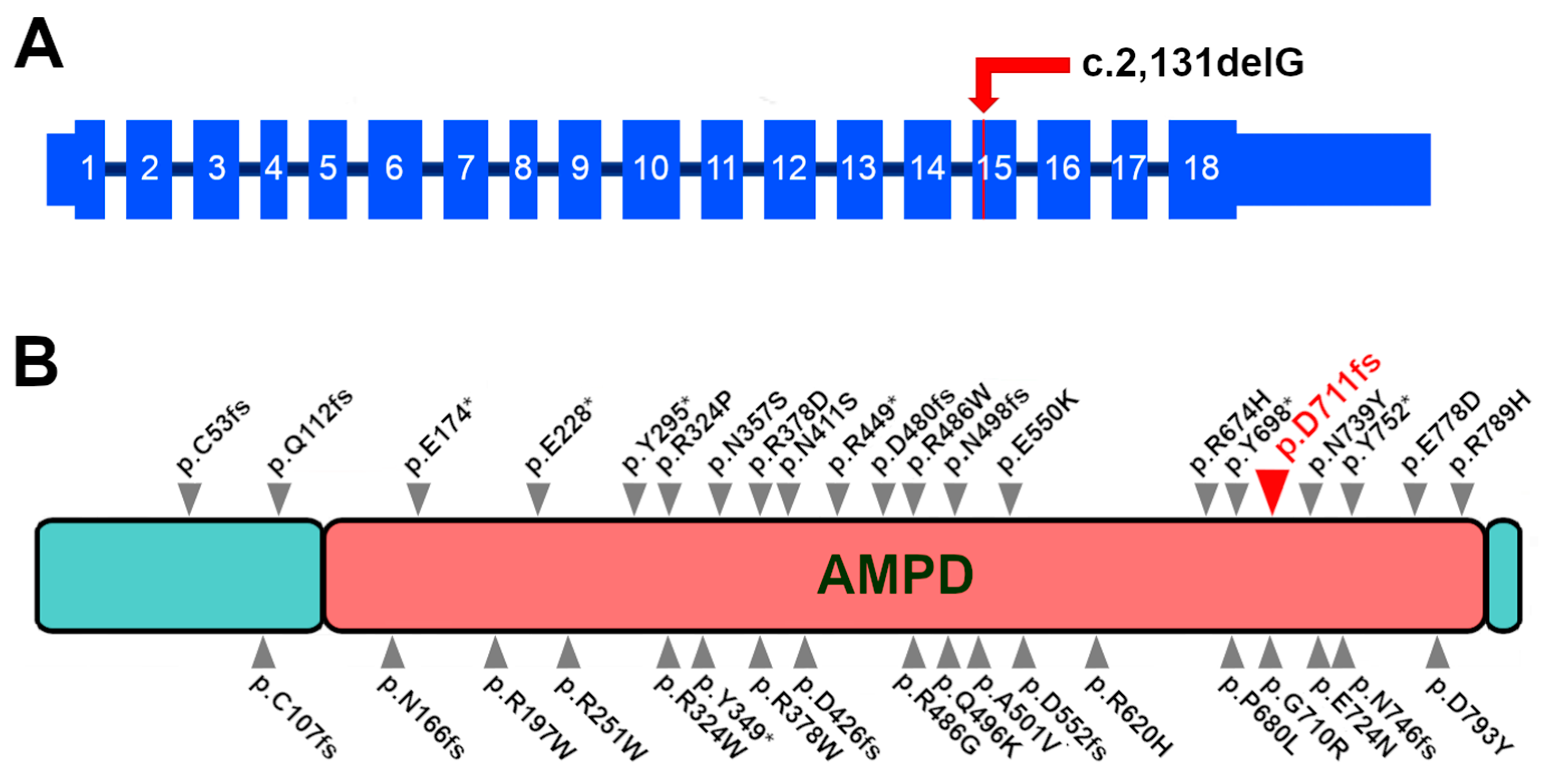
3.4. Variant Detection and Genotyping
3.5. Variant Position and Gene Expression in Mammals
3.6. Population Genetics and Affected Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Phan, T.M.L.; Zekveld-Vroon, R.C.; Leroy, B.P.; van den Born, L.I.; Hoyng, C.B.; Klaver, C.C.; Writing Committee for the Cone Disorders Study Group Consortium; Roosing, S.; Pott, J.-W.R.; et al. Clinical Course, Genetic Etiology, and Visual Outcome in Cone and Cone–Rod Dystrophy. Ophthalmology 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.C.; Traboulsi, E.I. Leber congenital amaurosis: Clinical correlations with genotypes, gene therapy trials update, and future directions. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2009, 13, 587–592. [Google Scholar] [CrossRef]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Mohnen, D.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. (Eds.) Essentials of Glycobiology, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022. [Google Scholar]
- Nita, D.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016, 18, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. S5), v4–v12. [Google Scholar] [CrossRef]
- Imanaka, T. Biogenesis and Function of Peroxisomes in Human Disease with a Focus on the ABC Transporter. Biol. Pharm. Bull. 2019, 42, 649–665. [Google Scholar] [CrossRef]
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef]
- Adams, N.A.; Awadein, A.; Toma, H.S. The Retinal Ciliopathies. Ophthalmic Genet. 2007, 28, 113–125. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Bardet-Biedl Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 171–174. [Google Scholar] [CrossRef]
- Valente, E.M.; Dallapiccola, B.; Bertini, E. Joubert syndrome and related disorders. Handb. Clin. Neurol. 2013, 113, 1879–1888. [Google Scholar] [CrossRef]
- Géléoc, G.G.; El-Amraoui, A. Disease mechanisms and gene therapy for Usher syndrome. Hear. Res. 2020, 394, 107932. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Senior-Loken Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Alstrom Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; You, Y. Editorial: Retinal Changes in Neurological Diseases. Front. Neurosci. 2021, 15, 813044. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Shi, H.; Oviatt, M.; Doustar, J.; Rentsendorj, A.; Fuchs, D.-T.; Sheyn, J.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Alzheimer’s Retinopathy: Seeing Disease in the Eyes. Front. Neurosci. 2020, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Fuchs, D.-T.; Sheyn, J.; Black, K.L.; Mirzaei, N.; Koronyo-Hamaoui, M. Retinal Vasculopathy in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 731614. [Google Scholar] [CrossRef]
- Lin, J.B.; Apte, R.S. Seeing Parkinson Disease in the Retina. JAMA Ophthalmol. 2021, 139, 189–190. [Google Scholar] [CrossRef]
- Chen, P.-C.; Chung, C.-C.; Cheng, Y.-Y.; Chen, W.-T.; Hong, C.-T.; Chan, L.; Chien, L.-N. Retinal Diseases and Parkinson Disease: A Population-Based Study. Front. Neurosci. 2021, 15, 679092. [Google Scholar] [CrossRef]
- Kolesnikova, M.; de Carvalho, J.R.L.; Oh, J.K.; Soucy, M.; Demirkol, A.; Kim, A.H.; Tsang, S.H.; Breazzano, M.P. Phenotypic Variability of Retinal Disease Among a Cohort of Patients With Variants in the CLN Genes. Investig. Opthalmol. Vis. Sci. 2023, 64, 23. [Google Scholar] [CrossRef]
- Mole, S.E. The Genetic Spectrum of Human Neuronal Ceroid-lipofuscinoses. Brain Pathol. 2004, 14, 70–76. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’brien, D.P.; Sohar, I.; Lobel, P.; Coates, J.R.; Khan, S.; Johnson, G.C.; Giger, U.; Johnson, G.S. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 2006, 89, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Coates, J.R.; Cooper, J.J.; O’Brien, D.P.; Jeong, M.; Narfstro¨m, K. Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. Investig. Opthalmol. Vis. Sci. 2008, 49, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Winkler, P.A.; Occelli, L.M.; Petersen-Jones, S.M. Large Animal Models of Inherited Retinal Degenerations: A Review. Cells 2020, 9, 882. [Google Scholar] [CrossRef]
- Miyadera, K. Mapping of Canine Models of Inherited Retinal Diseases. Adv. Exp. Med. Biol. 2018, 1074, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Khanna, H.; Punzo, C. Advances in gene therapy for diseases of the eye. Hum. Gene Ther. 2016, 27, 563–579. [Google Scholar] [CrossRef]
- Aguirre, G.D. Concepts and strategies in retinal gene therapy. Investig. Opthalmol. Vis. Sci. 2017, 58, 5399–5411. [Google Scholar] [CrossRef]
- Guziewicz, K.E.; Cideciyan, A.V.; Beltran, W.A.; Komáromy, A.M.; Dufour, V.L.; Swider, M.; Iwabe, S.; Sumaroka, A.; Kendrick, B.T.; Ruthel, G.; et al. BEST1 gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure. Proc. Natl. Acad. Sci. USA 2018, 115, E2839–E2848. [Google Scholar] [CrossRef]
- Komáromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Miyadera, K.; Acland, G.M.; Aguirre, G.D. Genetic and phenotypic variations of inherited retinal diseases in dogs: The power of within- and across-breed studies. Mamm. Genome 2012, 23, 40–61. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Aleman, T.S.; Gu, D.; Pearce-Kelling, S.E.; Sumaroka, A.; Acland, G.M.; Aguirre, G.D. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2005, 102, 5233–5238. [Google Scholar] [CrossRef] [PubMed]
- Tricomi, D.; Moore, J. Canis Lupus Familiaris: Cirneco Dell’etna; Castel: Aicurzio, Italy, 2016. [Google Scholar]
- Cortellari, M.; Bionda, A.; Talenti, A.; Ceccobelli, S.; Attard, G.; Lasagna, E.; Crepaldi, P.; Liotta, L. Genomic variability of Cirneco dell’Etna and the genetic distance with other dog breeds. Ital. J. Anim. Sci. 2021, 20, 304–314. [Google Scholar] [CrossRef]
- Puurunen, J.; Ottka, C.; Salonen, M.; Niskanen, J.E.; Lohi, H. Age, breed, sex and diet influence serum metabolite profiles of 2000 pet dogs. R. Soc. Open Sci. 2022, 9, 211642. [Google Scholar] [CrossRef] [PubMed]
- Talenti, A.; Dreger, D.L.; Frattini, S.; Polli, M.; Marelli, S.; Harris, A.C.; Liotta, L.; Cocco, R.; Hogan, A.N.; Bigi, D.; et al. Studies of modern Italian dog populations reveal multiple patterns for domestic breed evolution. Ecol. Evol. 2018, 8, 2911–2925. [Google Scholar] [CrossRef]
- Sinnwell, J.P.; Therneau, T.M.; Schaid, D.J. The kinship2 R Package for Pedigree Data. Hum. Hered. 2014, 78, 91–93. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.G.; Dreger, D.L.; Rimbault, M.; Davis, B.W.; Mullen, A.B.; Carpintero-Ramirez, G.; Ostrander, E.A. Genomic Analyses Reveal the Influence of Geographic Origin, Migration, and Hybridization on Modern Dog Breed Development. Cell Rep. 2017, 19, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Meadows, J.R.S.; Kidd, J.M.; Wang, G.-D.; Parker, H.G.; Schall, P.Z.; Bianchi, M.; Christmas, M.J.; Bougiouri, K.; Buckley, R.M.; Hitte, C.; et al. Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture. Genome Biol. 2023, 24, 187. [Google Scholar] [CrossRef]
- Ye, S.-S.; Tang, Y.; Song, J.-T. ATP and Adenosine in the Retina and Retinal Diseases. Front. Pharmacol. 2021, 12, 654445. [Google Scholar] [CrossRef]
- Barth, P.G. Pontocerebellar hypoplasias: An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993, 15, 411–422. [Google Scholar] [CrossRef]
- Maricich, S.M.; Aqeeb, K.A.; Moayedi, Y.; Mathes, E.L.; Patel, M.S.; Chitayat, D.; Lyon, G.; Leroy, J.G.; Zoghbi, H.Y. Pontocerebellar hypoplasia: Review of classification and genetics, and exclusion of several genes known to be important for cerebellar development. J. Child Neurol. 2011, 26, 288–294. [Google Scholar] [CrossRef]
- Kortüm, F.; Jamra, R.A.; Alawi, M.; Berry, S.A.; Borck, G.; Helbig, K.L.; Tang, S.; Huhle, D.; Korenke, G.C.; Hebbar, M.; et al. Clinical and genetic spectrum of AMPD2-related pontocerebellar hypoplasia type 9. Eur. J. Hum. Genet. 2018, 26, 695–708. [Google Scholar] [CrossRef]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D.; Naviaux, R.K.; Van Vleet, J.; Fenstermaker, A.G.; Silhavy, J.L.; et al. AMPD2 Regulates GTP Synthesis and Is Mutated in a Potentially Treatable Neurodegenerative Brainstem Disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef]
- Van Cauter, S.; Severino, M.; Ammendola, R.; Van Berkel, B.; Vavro, H.; Hauwe, L.v.D.; Rumboldt, Z. Bilateral lesions of the basal ganglia and thalami (central grey matter)—Pictorial review. Neuroradiology 2020, 62, 1565–1605. [Google Scholar] [CrossRef] [PubMed]
- Kitao, Y.; Saito, T.; Watanabe, S.; Ohe, Y.; Takahashi, K.; Akaki, T.; Adachi, T.; Doi, S.; Yamanaka, K.; Murai, Y.; et al. The discovery of 3,3-dimethyl-1,2,3,4-tetrahydroquinoxaline-1-carboxamides as AMPD2 inhibitors with a novel mechanism of action. Bioorganic Med. Chem. Lett. 2023, 80, 129110. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005, 86, 25–33. [Google Scholar] [CrossRef]
- Niyadurupola, N.; Sidaway, P.; Ma, N.; Rhodes, J.D.; Broadway, D.C.; Sanderson, J. P2X7Receptor activation mediates retinal ganglion cell death in a human retina model of ischemic neurodegeneration. Investig. Opthalmol. Vis. Sci. 2013, 54, 2163–2170. [Google Scholar] [CrossRef] [PubMed]
- Notomi, S.; Hisatomi, T.; Kanemaru, T.; Takeda, A.; Ikeda, Y.; Enaida, H.; Kroemer, G.; Ishibashi, T. Critical involvement of extracellular atp acting on p2rx7 purinergic receptors in photoreceptor cell death. Am. J. Pathol. 2011, 179, 2798–2809. [Google Scholar] [CrossRef]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.-G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Namavar, Y.; Barth, P.G.; Kasher, P.R.; van Ruissen, F.; Brockmann, K.; Bernert, G.; Writzl, K.; Ventura, K.; Cheng, E.Y.; Ferriero, D.M.; et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 2011, 134, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Helmering, J.; Juan, T.; Li, C.M.; Chhoa, M.; Baron, W.; Gyuris, T.; Richards, W.G.; Turk, J.R.; Lawrence, J.; Cosgrove, P.A.; et al. A mutation in Ampd2 is associated with nephrotic syndrome and hypercholesterolemia in mice. Lipids Health Dis. 2014, 13, 167. [Google Scholar] [CrossRef] [PubMed]
- Toyama, K.; Morisaki, H.; Cheng, J.; Kawachi, H.; Shimizu, F.; Ikawa, M.; Okabe, M.; Morisaki, T. Proteinuria in AMPD2-deficient mice. Genes Cells 2012, 17, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Corbo, J.C.; Myers, C.A.; Lawrence, K.A.; Jadhav, A.P.; Cepko, C.L. A typology of photoreceptor gene expression patterns in the mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 12069–12074. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Barth, P.G.; Zerres, K. Pontocerebellar hypoplasia. Am. J. Med Genet. Part C Semin. Med Genet. 2014, 166, 173–183. [Google Scholar] [CrossRef]
- Maloy, S.R.; Hughes, K.T. Brenner’s Encyclopedia of Genetics; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Manifestation | Detection |
|---|---|
| Ocular findings | |
| Retinal degeneration | ~4 years |
| Cataracts | Unknown |
| Neurological findings | |
| Head bobbing and tremors | ~2–2.5 years |
| Atypical seizure activity | ~2.5 years |
| Signs of cervical pain | ~2 years |
| Chromosome | Start (bp) | End (bp) |
|---|---|---|
| chr4 | 30,472,934 | 33,342,556 |
| chr5 | 41,535,525 | 42,456,236 |
| chr6 | 13,160,052 | 78,103,814 |
| chr10 | 18,760,527 | 30,002,859 |
| chr20 | 16,053,617 | 20,940,104 |
| chr27 | 5,115,516 | 6,885,858 |
| chr34 | 32,724,176 | 39,490,788 |
| Breed and Availability of Retinal Phenotype | N of Dogs | Genotype | ||
|---|---|---|---|---|
| wt/wt | wt/Del | Del/Del | ||
| Cirneco Dell’etna—Cases | 2 | 0 | 0 | 2 |
| Cirneco Dell’etna—Unaffected | 33 | 30 | 3 * | 0 |
| Dog10k | 1204 | 1204 | 0 | 0 |
| Total | 1239 | 1234 | 3 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murgiano, L.; Niggel, J.K.; Benedicenti, L.; Cortellari, M.; Bionda, A.; Crepaldi, P.; Liotta, L.; Aguirre, G.K.; Beltran, W.A.; Aguirre, G.D. Frameshift Variant in AMPD2 in Cirneco dell’Etna Dogs with Retinopathy and Tremors. Genes 2024, 15, 238. https://doi.org/10.3390/genes15020238
Murgiano L, Niggel JK, Benedicenti L, Cortellari M, Bionda A, Crepaldi P, Liotta L, Aguirre GK, Beltran WA, Aguirre GD. Frameshift Variant in AMPD2 in Cirneco dell’Etna Dogs with Retinopathy and Tremors. Genes. 2024; 15(2):238. https://doi.org/10.3390/genes15020238
Chicago/Turabian StyleMurgiano, Leonardo, Jessica K. Niggel, Leontine Benedicenti, Matteo Cortellari, Arianna Bionda, Paola Crepaldi, Luigi Liotta, Geoffrey K. Aguirre, William A. Beltran, and Gustavo D. Aguirre. 2024. "Frameshift Variant in AMPD2 in Cirneco dell’Etna Dogs with Retinopathy and Tremors" Genes 15, no. 2: 238. https://doi.org/10.3390/genes15020238
APA StyleMurgiano, L., Niggel, J. K., Benedicenti, L., Cortellari, M., Bionda, A., Crepaldi, P., Liotta, L., Aguirre, G. K., Beltran, W. A., & Aguirre, G. D. (2024). Frameshift Variant in AMPD2 in Cirneco dell’Etna Dogs with Retinopathy and Tremors. Genes, 15(2), 238. https://doi.org/10.3390/genes15020238