
The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA and RNA Extraction, and Sequencing
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Repeat Sequence Analysis and MTPT Prediction
2.4. Phylogenetic Analysis
2.5. RNA Editing Site Identification and Validation
3. Results
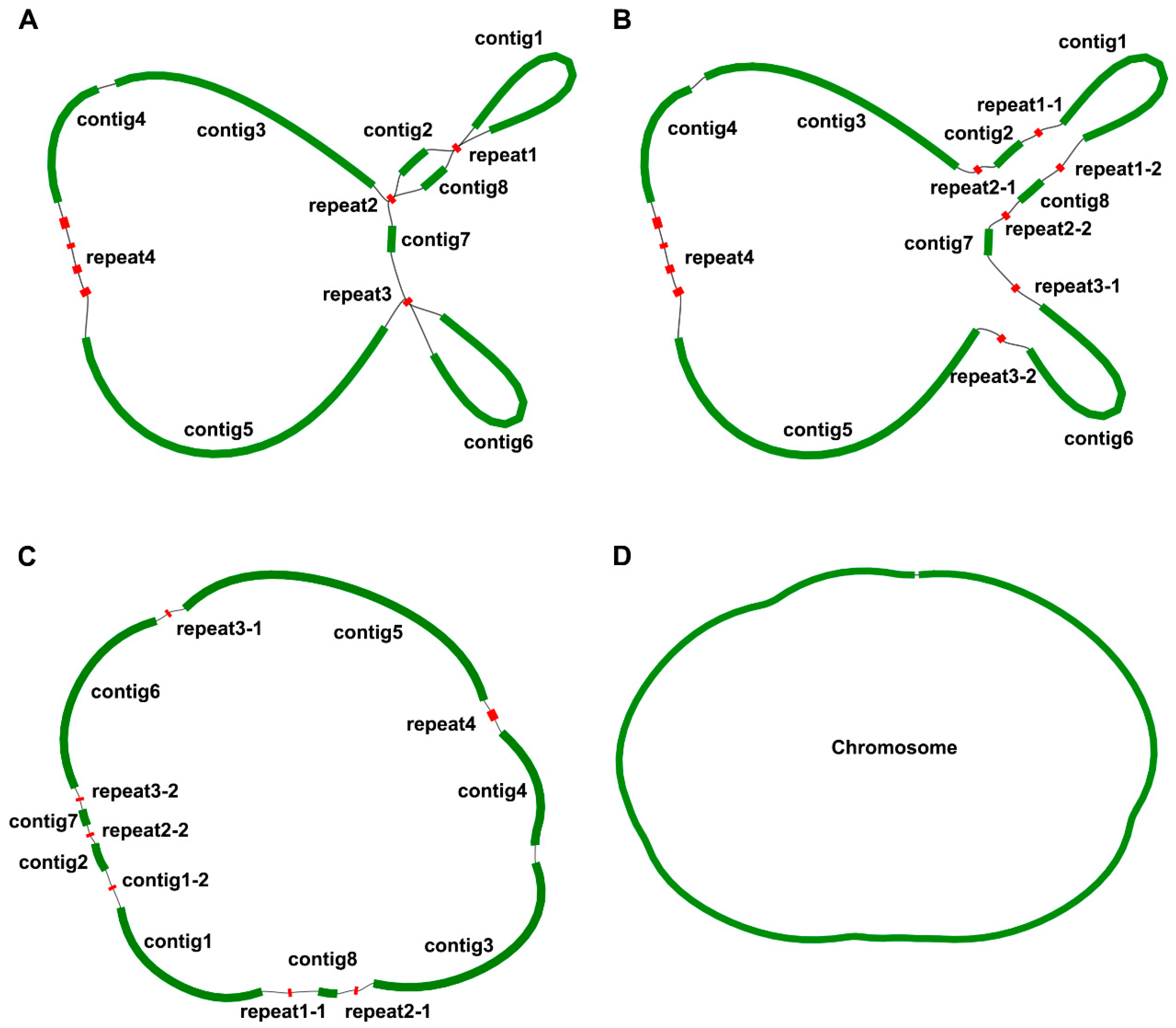
3.1. Elucidating Mitogenome Structure and Broad Genomic Features through Graph-Based Techniques
3.2. Genome Annotation
3.3. Repeat Element Analysis
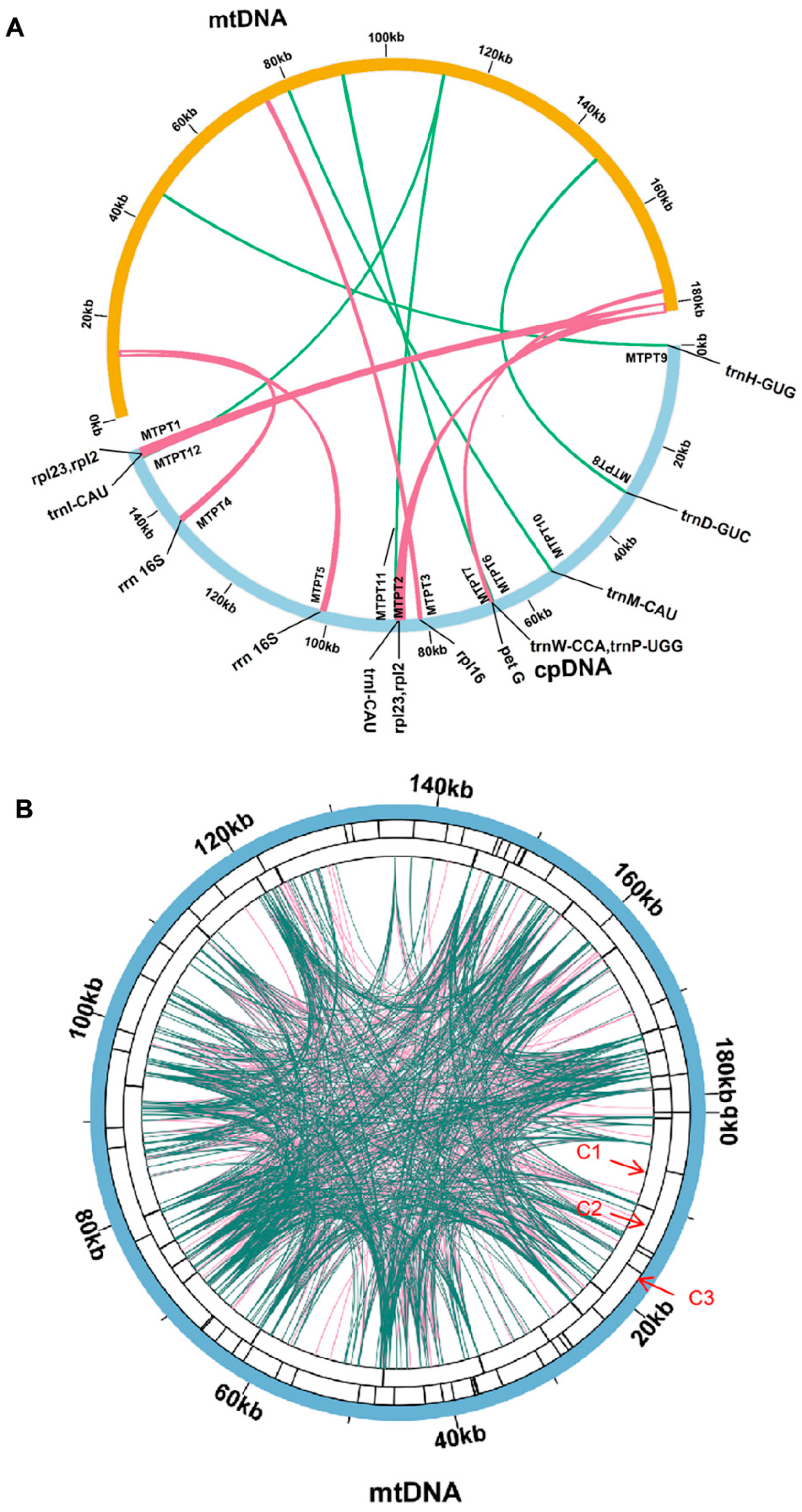
3.4. Mitochondrial Plastid DNAs Prediction
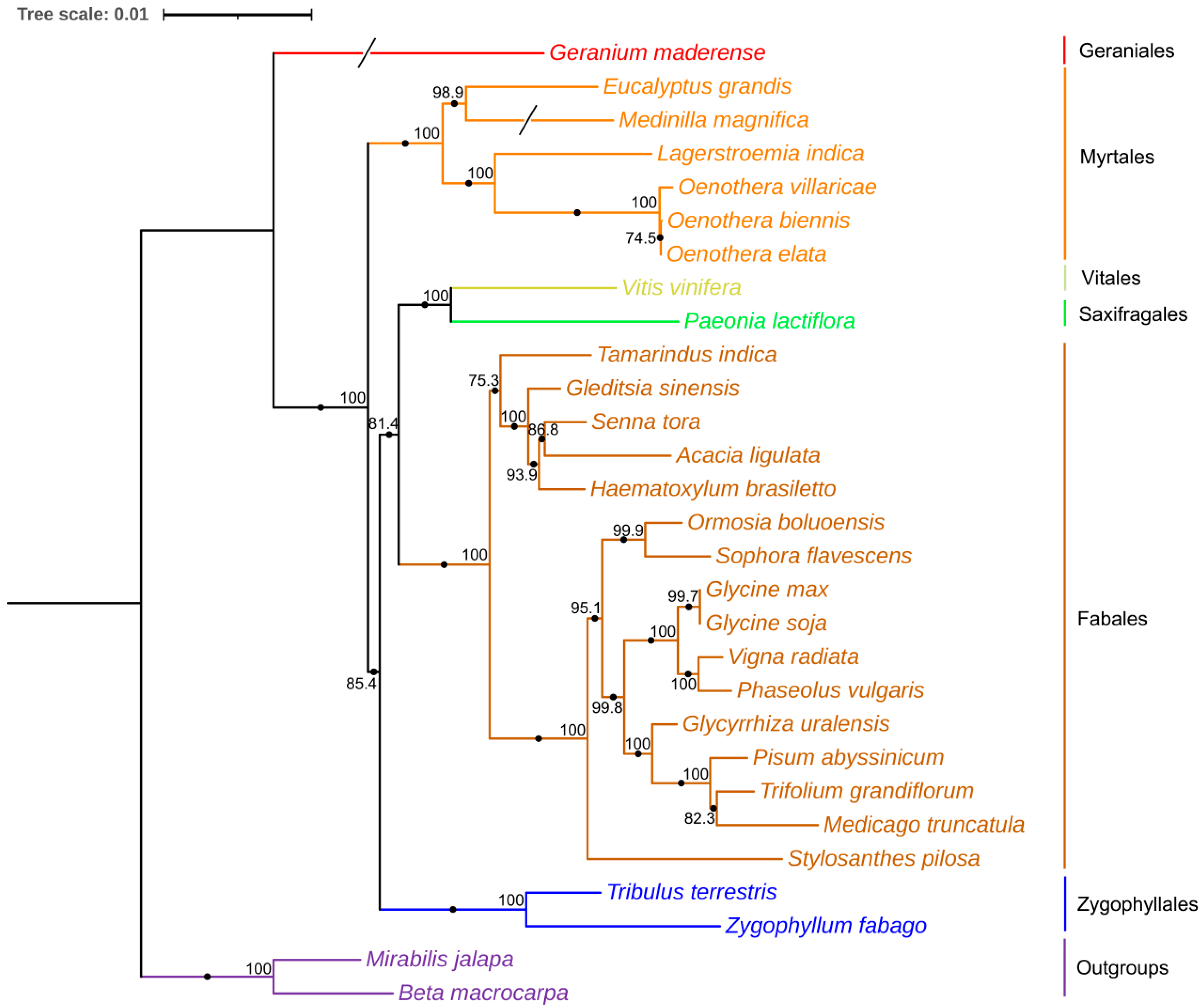
3.5. Phylogenetic Relationships
3.6. Prediction of RNA-Editing Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wang, Y.; He, J.; Wu, Z.; Huang, L.; Gao, M.; Li, M.; Feng, J. The Complete Mitogenome and Phylogeny Analysis of Pseudohemiculter Hainanensis (Boulenger, 1900) (Cyprinidae: Cultrinae). Mitochondrial DNA Part B 2022, 7, 2056–2059. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Q.; Ying, Z.; Ge, Y.; Cheng, R. Molecular Structure and Phylogenetic Analysis of Complete Chloroplast Genomes of Medicinal Species Paeonia Lactiflora from Zhejiang Province. Mitochondrial DNA Part B 2020, 5, 1077–1078. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Guo, L.; Li, Y.; Yang, H.; Hu, X.; Song, C.; Hou, X. Development and Characterization of Microsatellite Markers Based on the Chloroplast Genome of Tree Peony. Genes 2022, 13, 1543. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.-Y.; Chang, H.-W.; Lin, C.-F.; Liu, C.-J.; Hsieh, C.-F.; Horng, J.-T. Characterization of the Anti-Influenza Activity of the Chinese Herbal Plant Paeonia lactiflora. Viruses 2014, 6, 1861–1875. [Google Scholar] [CrossRef]
- Pan, H.-T.; Xi, Z.-Q.; Wei, X.-Q.; Wang, K. A Network Pharmacology Approach to Predict Potential Targets and Mechanisms of “Ramulus Cinnamomi (Cassiae)—Paeonia Lactiflora” Herb Pair in the Treatment of Chronic Pain with Comorbid Anxiety and Depression. Ann. Med. 2022, 54, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and Variable Rates of Repeat-Mediated Mitochondrial Genome Rearrangement in a Genus of Plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Ma, B.; Yu, J.; Wang, J.; Liu, C. De Novo Hybrid Assembly of the Salvia Miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. Int. J. Mol. Sci. 2022, 23, 14267. [Google Scholar] [CrossRef]
- Lower, S.E.; Dion-Côté, A.-M.; Clark, A.G.; Barbash, D.A. Special Issue: Repetitive DNA Sequences. Genes 2019, 10, 896. [Google Scholar] [CrossRef]
- Bi, C.; Qu, Y.; Hou, J.; Wu, K.; Ye, N.; Yin, T. Deciphering the Multi-Chromosomal Mitochondrial Genome of Populus Simonii. Front. Plant Sci. 2022, 13, 914635. [Google Scholar] [CrossRef]
- Muller, H.; Ogereau, D.; Da Lage, J.-L.; Capdevielle, C.; Pollet, N.; Fortuna, T.; Jeannette, R.; Kaiser, L.; Gilbert, C. Draft Nuclear Genome and Complete Mitogenome of the Mediterranean Corn Borer, Sesamia Nonagrioides, a Major Pest of Maize. G3 Genes|Genomes|Genet 2021, 11, jkab155. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, W.; Xu, J.; Li, M.; Zhang, Y. Chloroplast Genome Evolution and Species Identification of Styrax (Styracaceae). BioMed Res. Int. 2022, 2022, 5364094. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, Z.; Cai, D.; Song, L.; Bai, J. The Chloroplast Genome Sequence and Phylogenetic Analysis of Apocynum venetum L. PLoS ONE 2022, 17, e0261710. [Google Scholar] [CrossRef] [PubMed]
- Skuza, L.; Gastineau, R.; Sielska, A. The Complete Chloroplast Genome of Secale Sylvestre (Poaceae: Triticeae). J. Appl. Genet. 2022, 63, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Shearman, J.R.; Sonthirod, C.; Naktang, C.; Sangsrakru, D.; Yoocha, T.; Chatbanyong, R.; Vorakuldumrongchai, S.; Chusri, O.; Tangphatsornruang, S.; Pootakham, W. Assembly of the Durian Chloroplast Genome Using Long PacBio Reads. Sci. Rep. 2020, 10, 15980. [Google Scholar] [CrossRef]
- Suzuki, Y. Informatics for PacBio Long Reads. Adv. Exp. Med. Biol. 2019, 1129, 119–129. [Google Scholar] [CrossRef]
- Sheng, M.; She, J.; Xu, W.; Hong, Y.; Su, Z.; Zhang, X. HpeNet: Co-Expression Network Database for de Novo Transcriptome Assembly of Paeonia Lactiflora Pall. Front. Genet. 2020, 11, 570138. [Google Scholar] [CrossRef]
- Ren, X.; Shi, Y.; Xue, Y.; Xue, J.; Tian, Y.; Wang, S.; Zhang, X. Seed Proteomic Profiles of Three Paeonia Varieties and Evaluation of Peony Seed Protein as a Food Product. BioMed Res. Int. 2020, 2020, 5271296. [Google Scholar] [CrossRef]
- Štorchová, H.; Stone, J.D.; Sloan, D.B.; Abeyawardana, O.A.J.; Müller, K.; Walterová, J.; Pažoutová, M. Homologous Recombination Changes the Context of Cytochrome b Transcription in the Mitochondrial Genome of Silene Vulgaris KRA. BMC Genom. 2018, 19, 874. [Google Scholar] [CrossRef]
- Qu, Y.; Zhou, P.; Tong, C.; Bi, C.; Xu, L.A. Assembly and Analysis of the Populus Deltoides Mitochondrial Genome: The First Report of a Multicircular Mitochondrial Conformation for the Genus Populus. J. For. Res. 2023, 34, 717–733. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant Organellar RNA Editing: What 30 Years of Research Has Revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef]
- Swoboda, P.; Hohn, B.; Gal, S. Somatic Homologous Recombination in Planta: The Recombination Frequency Is Dependent on the Allelic State of Recombining Sequences and May Be Influenced by Genomic Position Effects. Mol. Genet. Genom. 1993, 237, 33–40. [Google Scholar] [CrossRef]
- Van Norden, M.; Falls, Z.; Mandloi, S.; Segal, B.; Baysal, B.; Samudrala, R.; Elkin, P.L. The Role of C-to-U RNA Editing in Human Biodiversity. bioRxiv 2023. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Q.; Yin, P. RNA Editing Machinery in Plant Organelles. Sci. China Life Sci. 2018, 61, 162–169. [Google Scholar] [CrossRef]
- Zanlungo, S.; Moenne, A.; Holuigue, L.; Jordana, X.; Quiñones, V. Splicing and Editing of Rps10 Transcripts in Potato Mitochondria. Curr. Genet. 1995, 27, 565–571. [Google Scholar] [CrossRef]
- Quinones, V.; Zanlungo, S.; Holuigue, L.; Litvak, S.; Jordana, X. The Cox1 Initiation Codon Is Created by RNA Editing in Potato Mitochondria. Plant Physiol. 1995, 108, 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, S.F.; Dias, S.G.; Hardouin, P.; Pereira, F.R.; Lejeune, B.; de Souza, A.P. Transcription of Succinate Dehydrogenase Subunit 4 (Sdh4) Gene in Potato: Detection of Extensive RNA Editing and Co-Transcription with Cytochrome Oxidase Subunit III (Cox3) Gene. Curr. Genet. 2002, 41, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Moreira, J.D.; Smith, K.K.; Fetterman, J.L. The Mighty NUMT: Mitochondrial DNA Flexing Its Code in the Nuclear Genome. Biomolecules 2023, 13, 753. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, M.L.; Yuyama, P.M.; Sartori, D.; Fungaro, M.H.P.; Vanzela, A.L.L. Occurrence and Chromosome Distribution of Retroelements and NUPT Sequences in Copaifera Langsdorffii Desf. (Caesalpinioideae). Chromosom. Res. 2010, 18, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Hatmaker, E.A.; Wadl, P.A.; Rinehart, T.A.; Carroll, J.; Lane, T.S.; Trigiano, R.N.; Staton, M.E.; Schilling, E.E. Complete chloroplast genome comparisons for Pityopsis (Asteraceae). PLoS ONE 2020, 15, e0241391. [Google Scholar] [CrossRef]
- Gandini, C.L.; Sanchez-Puerta, M.V. Foreign Plastid Sequences in Plant Mitochondria are Frequently Acquired Via Mitochondrion-to-Mitochondrion Horizontal Transfer. Sci. Rep. 2017, 7, 43402. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Wu, Z. History of Plastid DNA Insertions Reveals Weak Deletion and AT Mutation Biases in Angiosperm Mitochondrial Genomes. Genome Biol. Evol. 2014, 6, 3210–3221. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Li, J.; Zhao, Q.; Liang, Y.; Yu, J. Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes 2021, 12, 1970. [Google Scholar] [CrossRef]
- Lv, S.; Cheng, S.; Wang, Z.; Li, S.; Jin, X.; Lan, L.; Yang, B.; Yu, K.; Ni, X.; Li, N.; et al. Draft Genome of the Famous Ornamental Plant Paeonia suffruticosa. Ecol. Evol. 2020, 10, 4518–4530. [Google Scholar] [CrossRef]
- Adamovic, D.; Djalovic, I.; Mitrovic, P.; Kojic, S.; Pivic, R.; Josic, D. First Report on Natural Infection of Paeonia tenuifolia by ‘Candidatus Phytoplasma Solani’ in Serbia. Plant Dis. 2014, 98, 565. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1962, pp. 1–14. ISBN 978-1-4939-9172-3. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The Alternative Reality of Plant Mitochondrial DNA: One Ring Does Not Rule Them All. PLOS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an Integrated Plastome Sequence Annotator and Analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGVIEW: A Package for Visualizing Detailed Chloroplast Genome Structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep Representation Learning for Predicting C-to-U RNA Editing in Plant Mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X.; Wu, K.; Pan, J.; Long, H.; Yan, Y. Characterization of Simple Sequence Repeats (SSRs) in Ciliated Protists Inferred by Comparative Genomics. Microorganisms 2020, 8, 662. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Xing, J.; Nie, T.; Su, A.; Zhang, R.; Zhao, Y.; Song, W.; Zhao, J. Comparative Analysis of Mitochondrial Genomes of Maize CMS-S Subtypes Provides New Insights into Male Sterility Stability. BMC Plant Biol. 2022, 22, 469. [Google Scholar] [CrossRef]
- Balzano, E.; Pelliccia, F.; Giunta, S. Genome (in)Stability at Tandem Repeats. Semin. Cell Dev. Biol. 2021, 113, 97–112. [Google Scholar] [CrossRef]
- Mehrotra, S.; Goyal, V. Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genom. Proteom. Bioinform. 2014, 12, 164–171. [Google Scholar] [CrossRef]
- Wang, X.-C.; Chen, H.; Yang, D.; Liu, C. Diversity of Mitochondrial Plastid DNAs (MTPTs) in Seed Plants. Mitochondrial DNA Part A 2018, 29, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Massenet, O.; Dome, A.-M.; Briat, J.-F.; Mache, R. Expression of the Rpl23, Rpl2 and Rps19 Genes in Spinach Chloroplasts. Nucleic Acids Res. 1988, 16, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Ong, H.C.; Palmer, J.D. Mitochondrial Gene Transfer in Pieces: Fission of the Ribosomal Protein Gene Rpl2 and Partial or Complete Gene Transfer to the Nucleus. Mol. Biol. Evol. 2001, 18, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Haberhausen, G.; Zetsche, K.; Bömmer, D. A Large Deletion in the Plastid DNA of the Holoparasitic Flowering Plant Cuscuta Reflexa Concerning Two Ribosomal Proteins (Rpl2, Rpl23), One Transfer RNA (trnI) and an ORF 2280 Homologue. Curr. Genet. 1993, 24, 171–176. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Liu, Y.; Shi, J.; Li, W.; Suo, Z. Chloroplast Phylogenomics and Divergence Times of Lagerstroemia (Lythraceae). BMC Genom. 2021, 22, 434. [Google Scholar] [CrossRef] [PubMed]
- Filipenko, E.A.; Sidorchuk, Y.V.; Deineko, E.V. Spontaneous spectinomycin resistance mutations of the chloroplast rrn16 gene in Daucus carota callus lines. Genetika 2011, 47, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.-N.; Wu, C.-S.; Ye, L.-J.; Mo, Z.-Q.; Liu, J.; Chang, Y.-W.; Li, D.-Z.; Chaw, S.-M.; Gao, L.-M. Prevalence of Isomeric Plastomes and Effectiveness of Plastome Super-Barcodes in Yews (Taxus) Worldwide. Sci. Rep. 2019, 9, 2773. [Google Scholar] [CrossRef]
- Brennicke, A.; Marchfelder, A.; Binder, S. RNA Editing. FEMS Microbiol. Rev. 1999, 23, 297–316. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Han, F.; Qu, Y.; Chen, Y.; Xu, L.; Bi, C. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Salix Wilsonii Using PacBio HiFi Sequencing. Front. Plant Sci. 2022, 13, 1031769. [Google Scholar] [CrossRef]
- Modi, A.; Vai, S.; Caramelli, D.; Lari, M. The Illumina Sequencing Protocol and the NovaSeq 6000 System. In Bacterial Pangenomics; Methods in Molecular Biology; Mengoni, A., Bacci, G., Fondi, M., Eds.; Springer: New York, NY, USA, 2021; Volume 2242, pp. 15–42. ISBN 978-1-07-161098-5. [Google Scholar]
- Yuan, J.; Jiang, S.; Jian, J.; Liu, M.; Yue, Z.; Xu, J.; Li, J.; Xu, C.; Lin, L.; Jing, Y.; et al. Genomic Basis of the Giga-Chromosomes and Giga-Genome of Tree Peony Paeonia Ostii. Nat. Commun. 2022, 13, 7328. [Google Scholar] [CrossRef]
- Giudice, C.L.; Hernández, I.; Ceci, L.R.; Pesole, G.; Picardi, E. RNA Editing in Plants: A Comprehensive Survey of Bioinformatics Tools and Databases. Plant Physiol. Biochem. 2019, 137, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, S.; Kumar, N.; Rana, R.S.; Sharma, P.; Kumar, P.; Rani, M. Morpho-Molecular Genetic Diversity and Population Structure Analysis in Garden Pea (Pisum sativum L.) Genotypes Using Simple Sequence Repeat Markers. PLoS ONE 2022, 17, e0273499. [Google Scholar] [CrossRef] [PubMed]
- Bedbrook, J.R.; Kolodner, R.; Bogorad, L. Zea Mays Chloroplast Ribosomal RNA Genes Are Part of a 22,000 Base Pair Inverted Repeat. Cell 1977, 11, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Long Terminal Repeat Retrotransposons of Oryza Sativa—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12372141/ (accessed on 29 December 2023).
- Vilela, M.d.M.; Del-Bem, L.-E.; Van Sluys, M.-A.; de Setta, N.; Kitajima, J.P.; Cruz, G.M.Q.; Sforça, D.A.; de Souza, A.P.; Ferreira, P.C.G.; Grativol, C.; et al. Analysis of Three Sugarcane Homo/Homeologous Regions Suggests Independent Polyploidization Events of Saccharum officinarum and Saccharum spontaneum. Genome Biol. Evol. 2017, 9, 266–278. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name | Length (bp) | Chromosomes | Bifurcation Structures |
|---|---|---|---|---|
| 1 | contig1 | 31,787 | Chromosome1 | No |
| 2 | contig2 | 3211 | Chromosome1 | No |
| 3 | contig3 | 35,924 | Chromosome1 | No |
| 4 | contig4 | 16,986 | Chromosome1 | No |
| 5 | contig5 | 51,364 | Chromosome1 | No |
| 6 | contig6 | 31,950 | Chromosome1 | No |
| 7 | contig7 | 2735 | Chromosome1 | No |
| 8 | contig8 | 4860 | Chromosome1 | No |
| 9 | repeat1-1 | 224 | Chromosome1 | Yes |
| 10 | repeat1-2 | 224 | Chromosome1 | Yes |
| 11 | repeat2-1 | 181 | Chromosome1 | Yes |
| 12 | repeat2-2 | 181 | Chromosome1 | Yes |
| 13 | repeat3-1 | 189 | Chromosome1 | Yes |
| 14 | repeat3-2 | 189 | Chromosome1 | Yes |
| 15 | repeat4 | 1683 | Chromosome1 | No |
| Group of Genes | Name of Genes |
|---|---|
| ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| NADH dehydrogenase | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 |
| Cytochrome b | cob |
| Cytochrome oxidase | cox1, cox2, cox3 |
| Maturases | matR |
| Protein transport subunit | mttB |
| Other genes | ccmFC, ccmB, ccmC, ccmFN |
| Ribosomal protein large subunit | rpl5, rpl16 |
| Ribosomal protein small subunit | rps3, rps4, rps7, rps12, rps14 |
| Succinate dehydrogenase | sdh4 |
| Ribosomal RNA genes | rrn5, rrn18, rrn26 |
| Transfer RNA genes | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnM-CAU, trnH-GUG, trnK-UUU, trnN-GUU, trnP-UGG(×2), trnQ-UUG, trnS-GCU, trnW-CCA, trnY-GUA, trnS-UGA, trnM-CAU(×2) |
| Number | Alignment Length (bp) | Mitogenome | Cpgenome | MTPT Annotation | ||
|---|---|---|---|---|---|---|
| Start | End | Start | End | |||
| MTPT1 | 1683 | 181,688 | 180,006 | 150,737 | 152,397 | Complete (rpl23), Partial (rpl2) |
| MTPT2 | 1683 | 180,006 | 181,688 | 84,737 | 86,397 | Complete (rpl23), Partial (rpl2) |
| MTPT3 | 504 | 75,234 | 74,739 | 81,290 | 81,791 | Partial (rpl16) |
| MTPT4 | 886 | 13,207 | 12,349 | 135,451 | 136,315 | Partial (rrn16S) |
| MTPT5 | 886 | 12,349 | 13,207 | 100,819 | 101,683 | Partial (rrn16S) |
| MTPT6 | 408 | 177,268 | 177,662 | 66,634 | 37,018 | Complete (trnW-CCA, trnP-UGG) |
| MTPT7 | 212 | 91,080 | 91,288 | 66,378 | 66,583 | Complete (petG) |
| MTPT8 | 142 | 147,487 | 147,346 | 31,196 | 31,332 | Complete (trnD-GUC) |
| MTPT9 | 79 | 46,463 | 46,541 | 1 | 79 | Complete (trnH-GUG) |
| MTPT10 | 77 | 79,847 | 79,771 | 53,073 | 53,149 | Complete (trnM-CAU) |
| MTPT11 | 79 | 111,678 | 111,604 | 86,457 | 86,535 | Complete (trnI-CAU) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, P.; Ni, Y.; Li, J.; Lu, Q.; Liu, C.; Guo, J. The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome. Genes 2024, 15, 239. https://doi.org/10.3390/genes15020239
Tang P, Ni Y, Li J, Lu Q, Liu C, Guo J. The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome. Genes. 2024; 15(2):239. https://doi.org/10.3390/genes15020239
Chicago/Turabian StyleTang, Pan, Yang Ni, Jingling Li, Qianqi Lu, Chang Liu, and Jinlin Guo. 2024. "The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome" Genes 15, no. 2: 239. https://doi.org/10.3390/genes15020239
APA StyleTang, P., Ni, Y., Li, J., Lu, Q., Liu, C., & Guo, J. (2024). The Complete Mitochondrial Genome of Paeonia lactiflora Pall. (Saxifragales: Paeoniaceae): Evidence of Gene Transfer from Chloroplast to Mitochondrial Genome. Genes, 15(2), 239. https://doi.org/10.3390/genes15020239