
Transcriptome Analysis Provides Insights into Catalpol Biosynthesis in the Medicinal Plant Rehmannia glutinosa and the Functional Characterization of RgGES Genes
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Transcriptome Construction
2.3. Functional Annotation
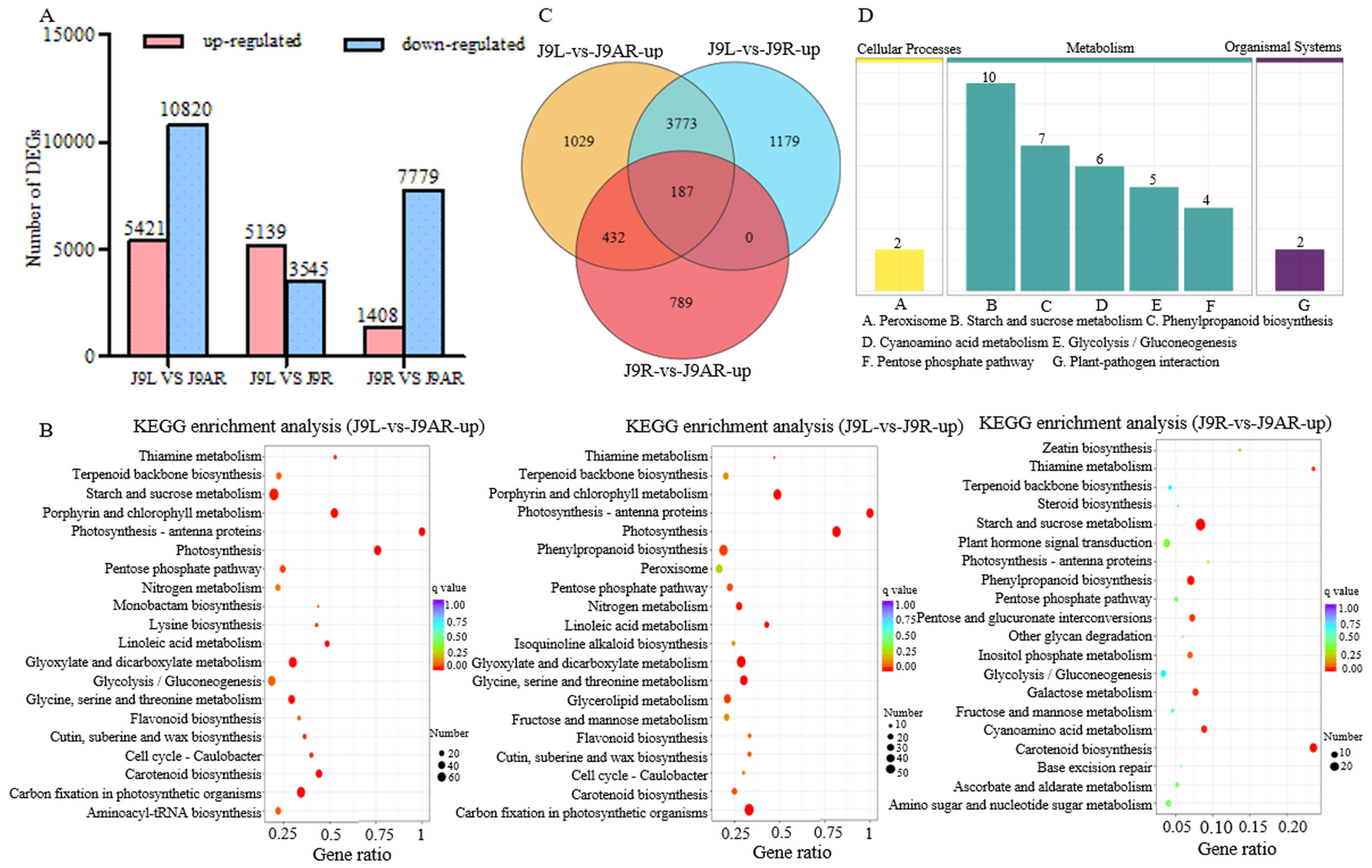
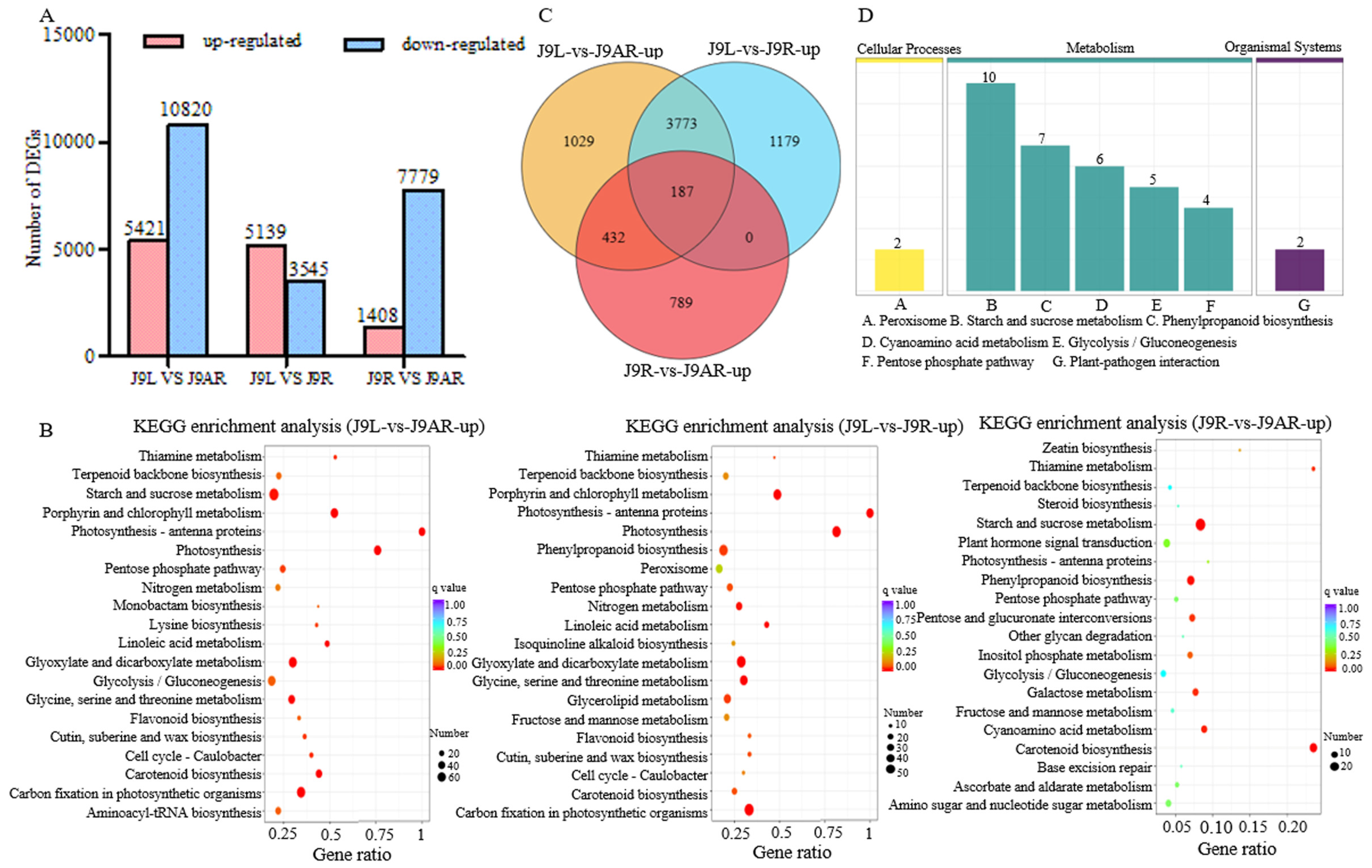
2.4. Identification of the Differentially Expressed Genes (DEGs)
2.5. HPLC Analysis
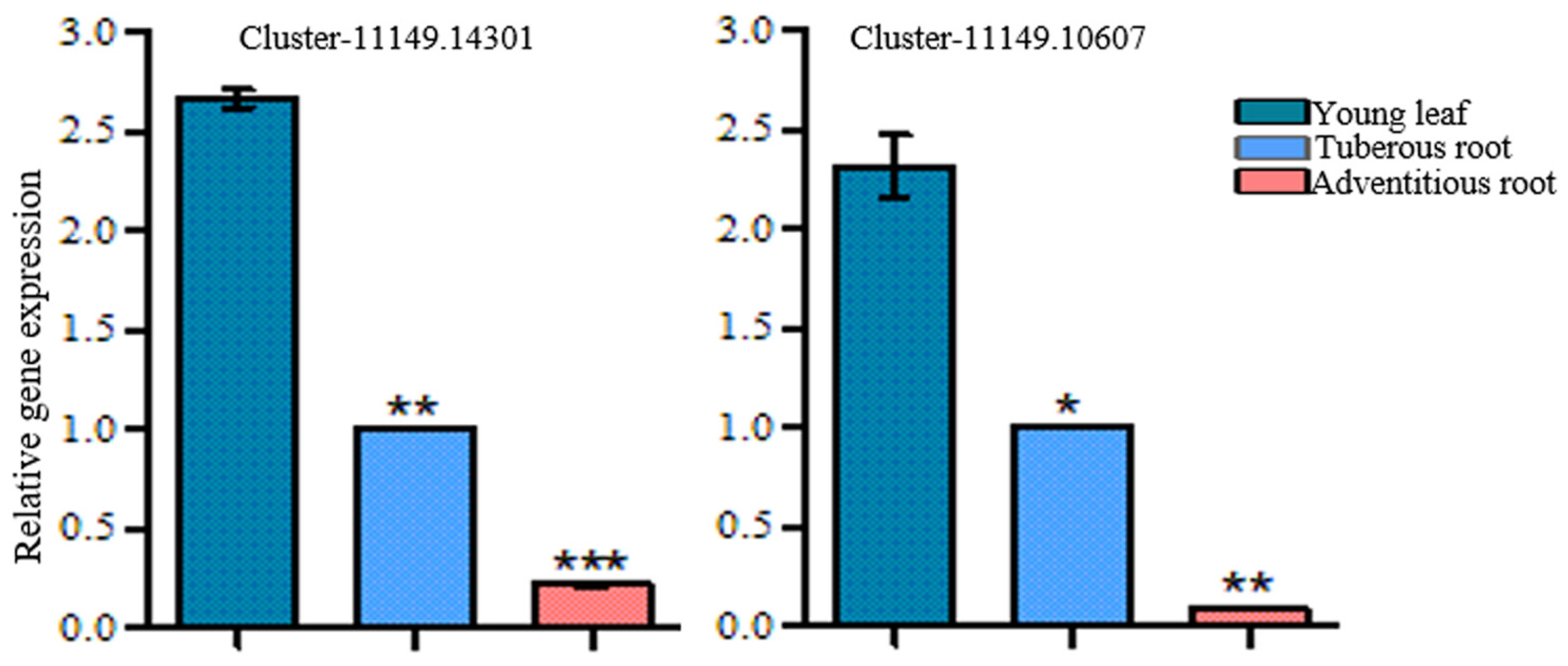
2.6. Gene Expression Analysis
2.7. Identification and Sequence Analysis of Candidate Geraniol Synthase Genes
2.8. Heterologous Expression of RgGES
2.9. Enzyme Assays
2.10. GC-MS Analysis
3. Results
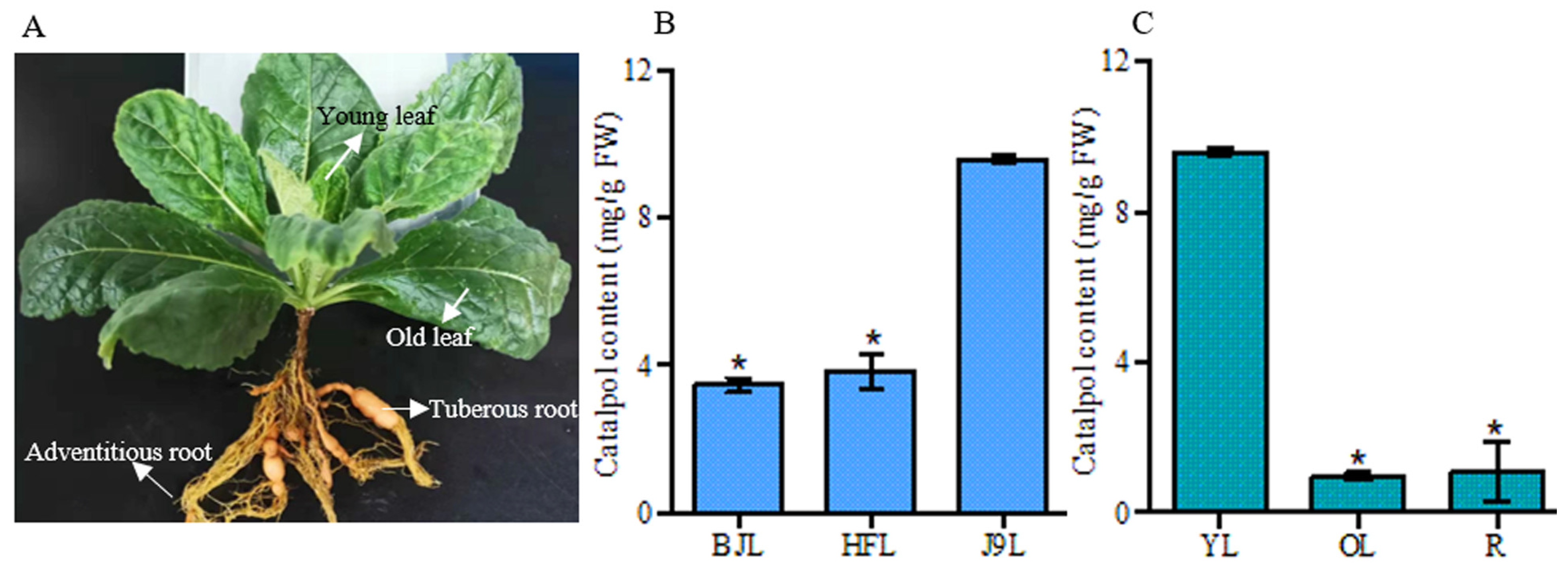
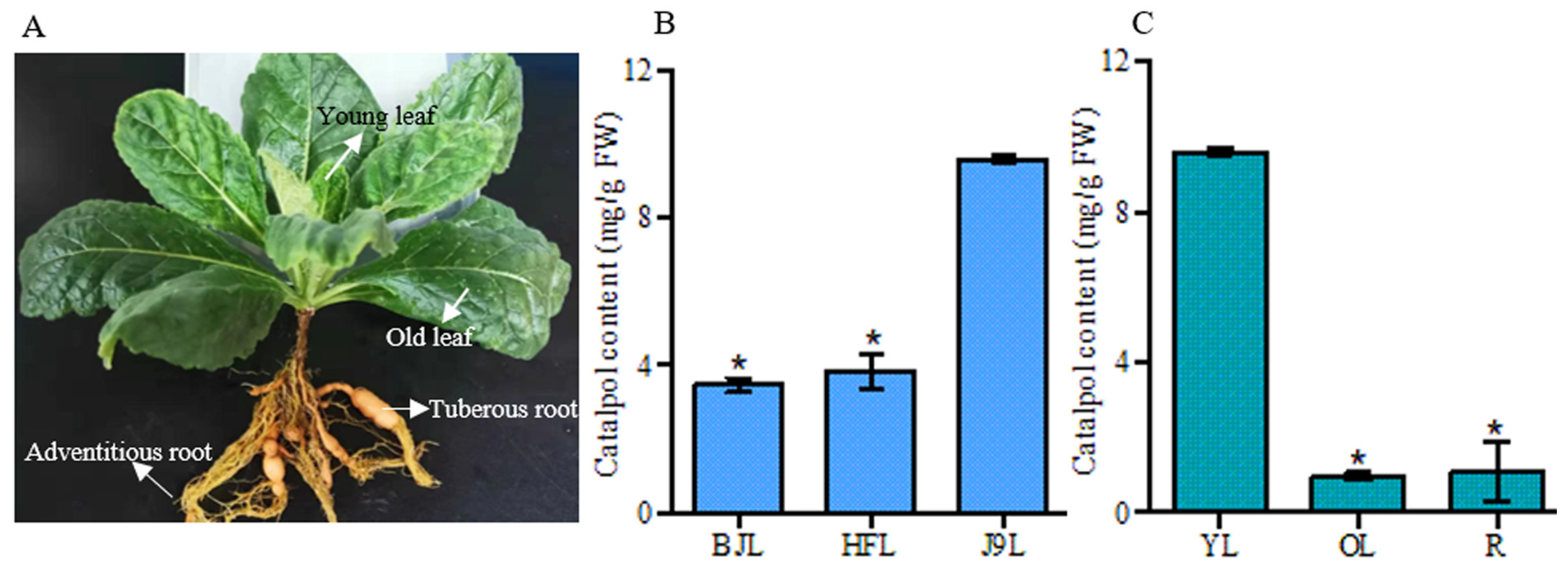
3.1. HPLC Analysis of Catalpol in R. glutinosa
3.2. Transcriptome Sequencing and De Novo Assembly
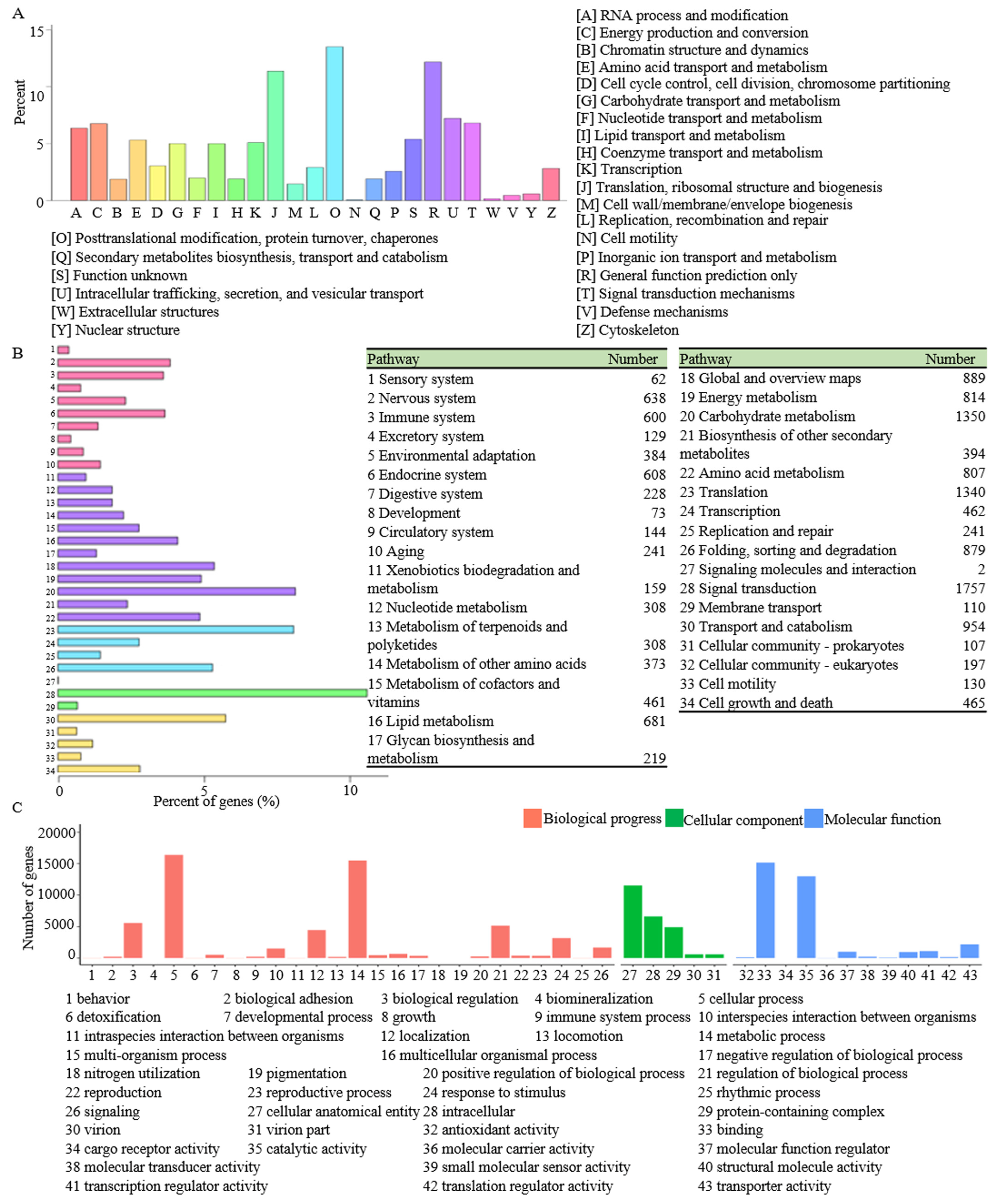
3.3. Functional Annotation and Classification of the R. glutinosa Unigenes
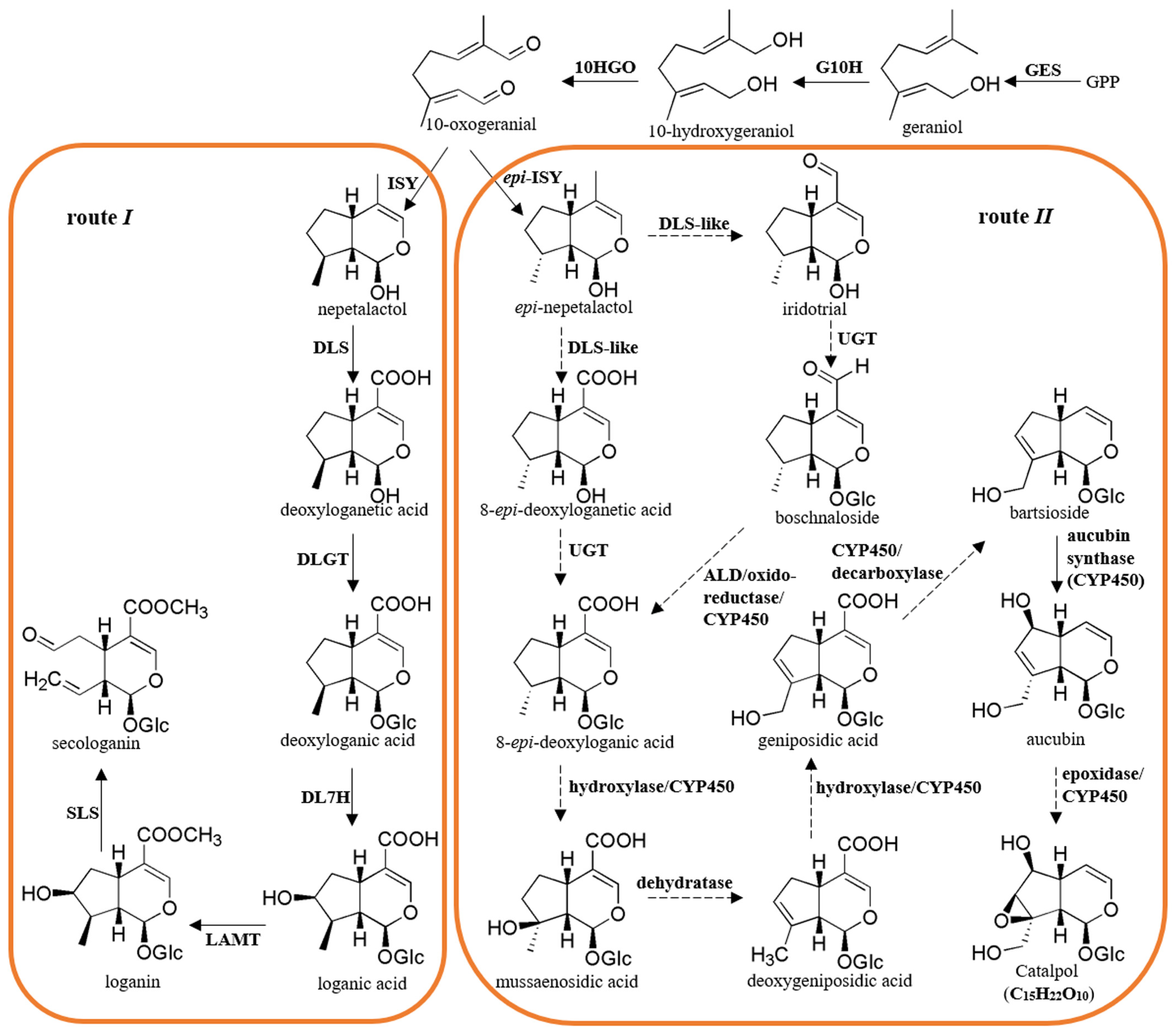
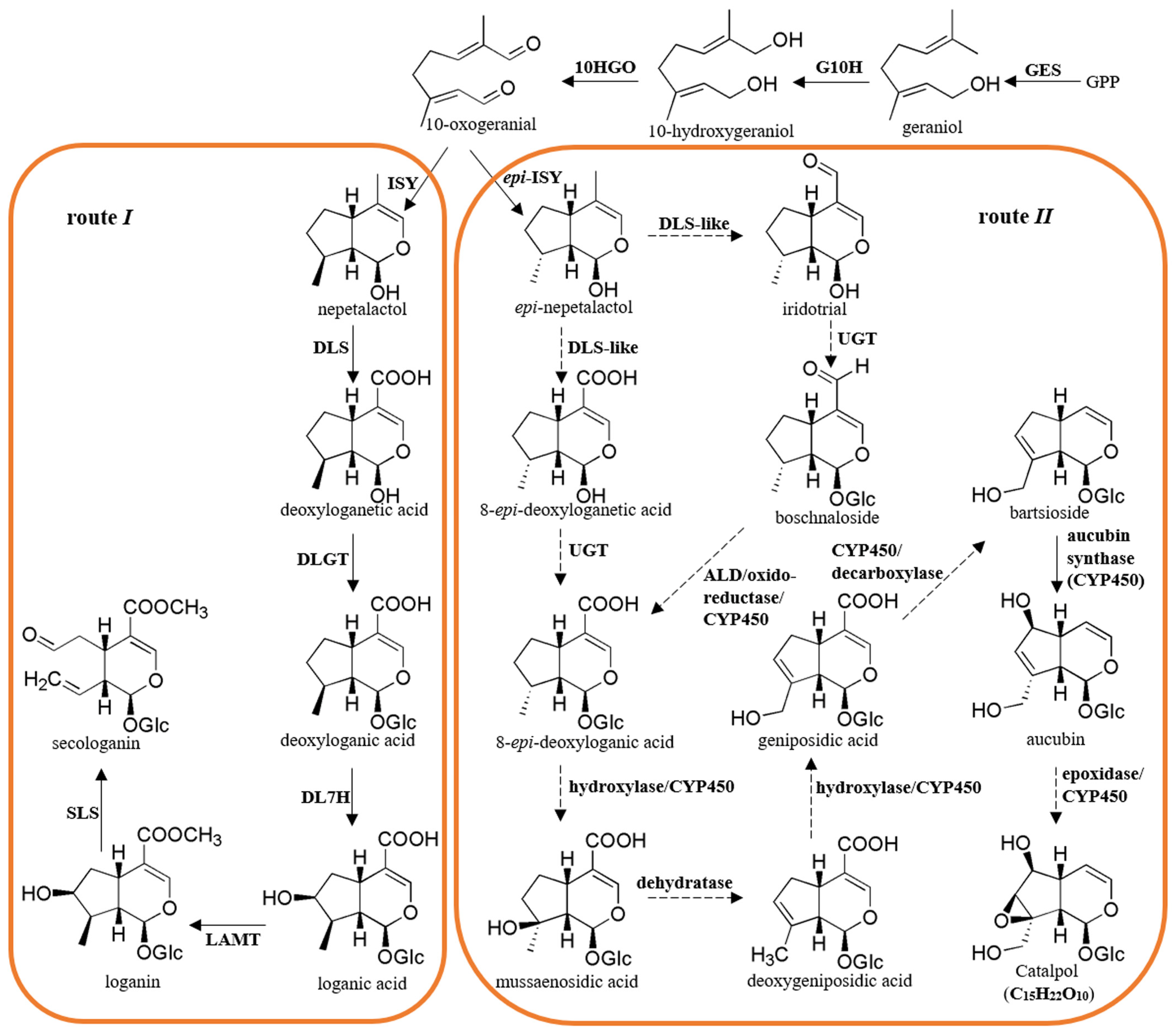
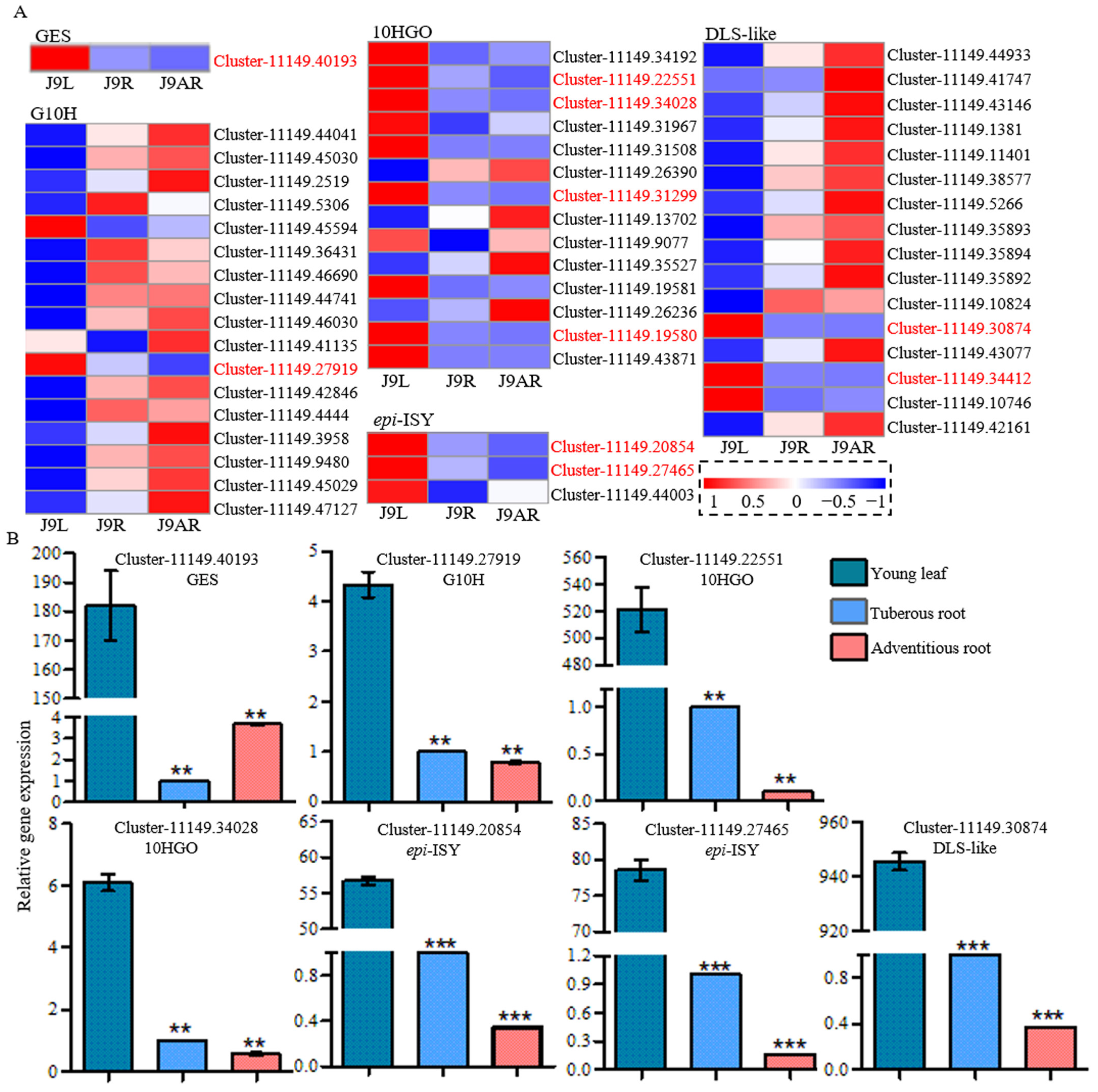
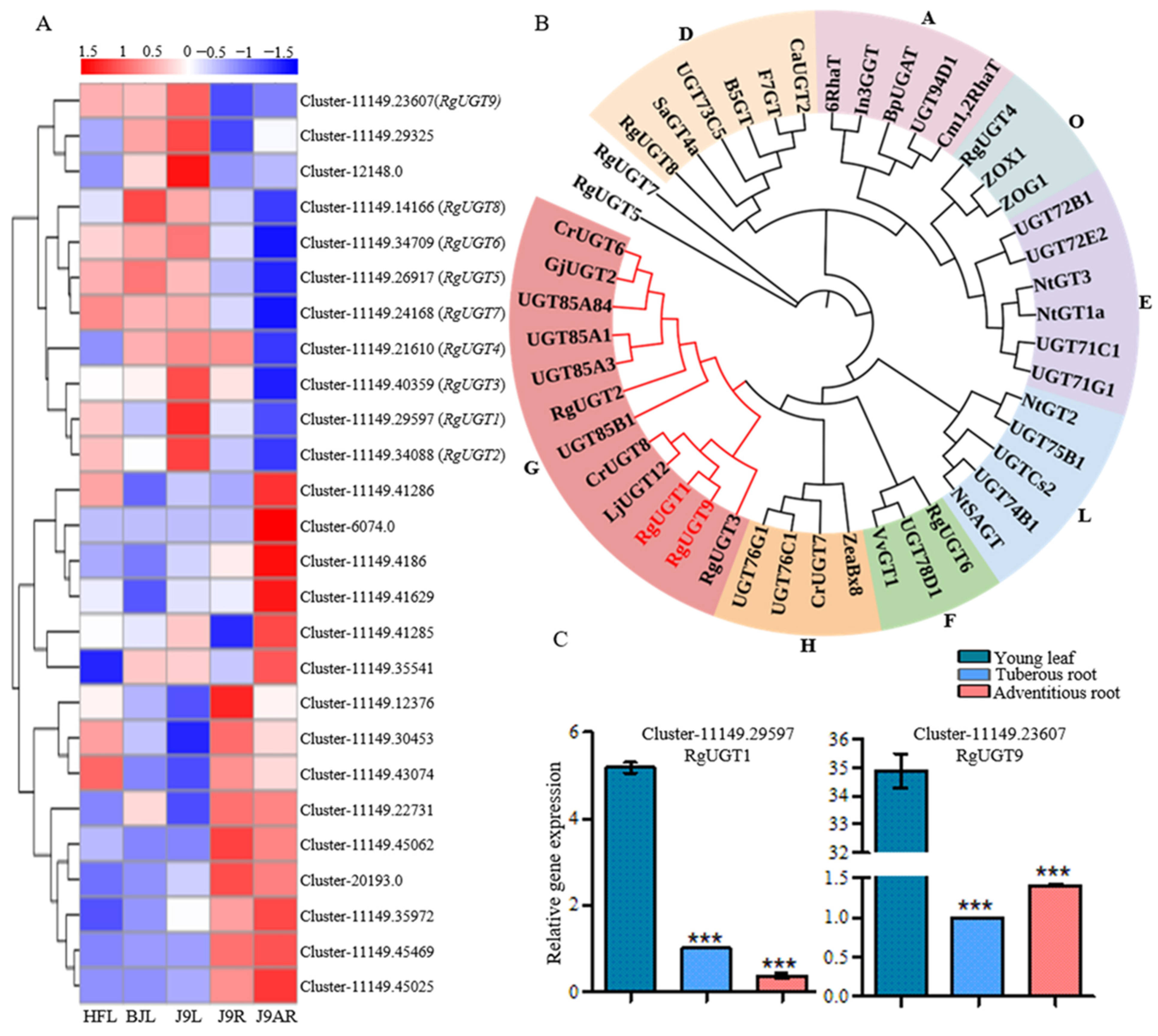
3.4. Identification of Putative Genes in the Pathway of Catalpol Biosynthesis
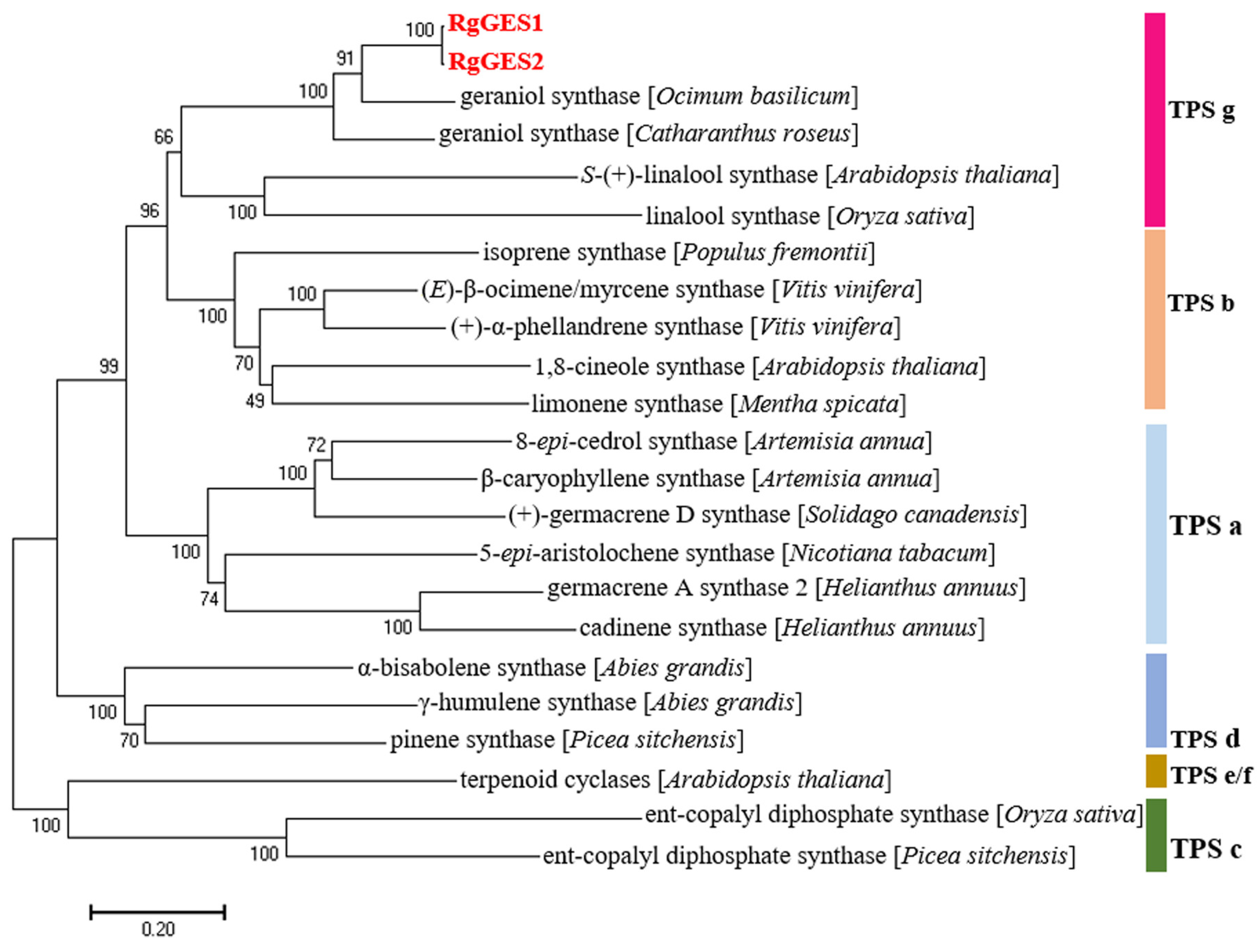
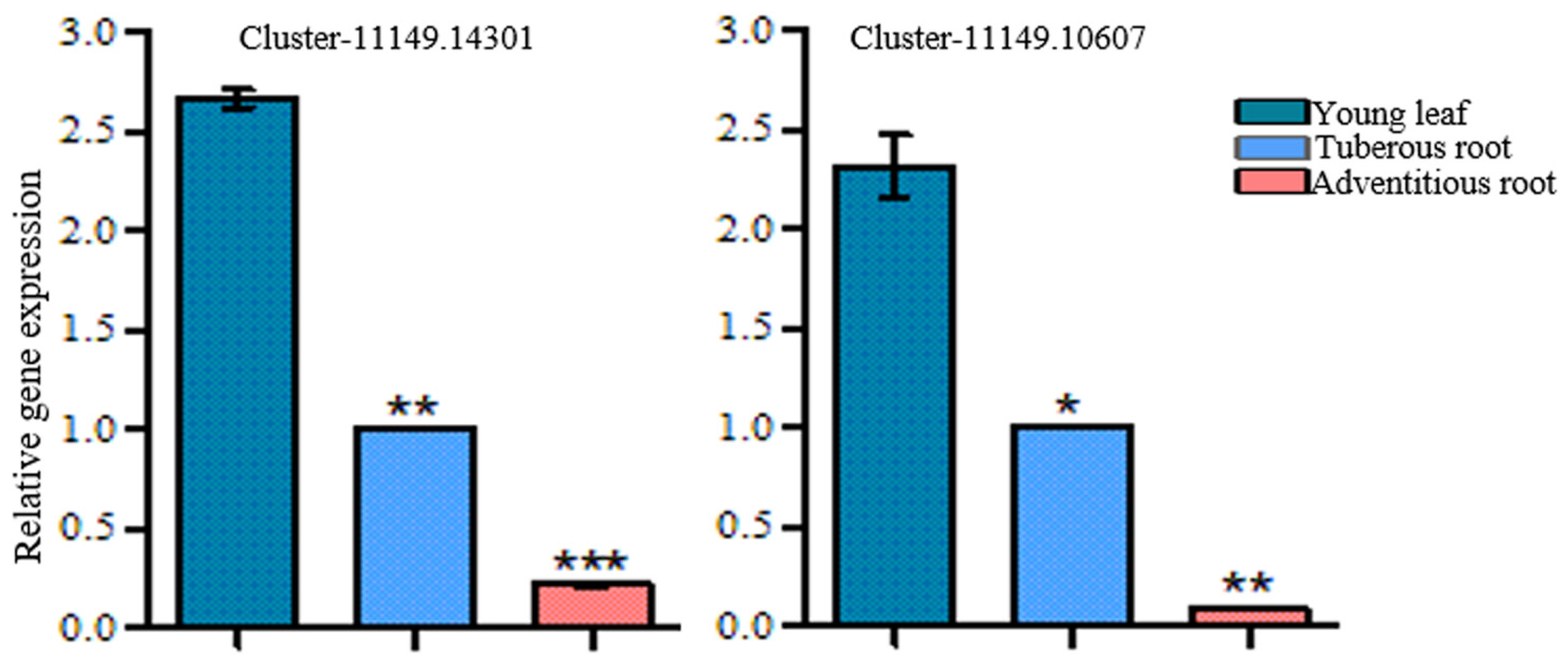
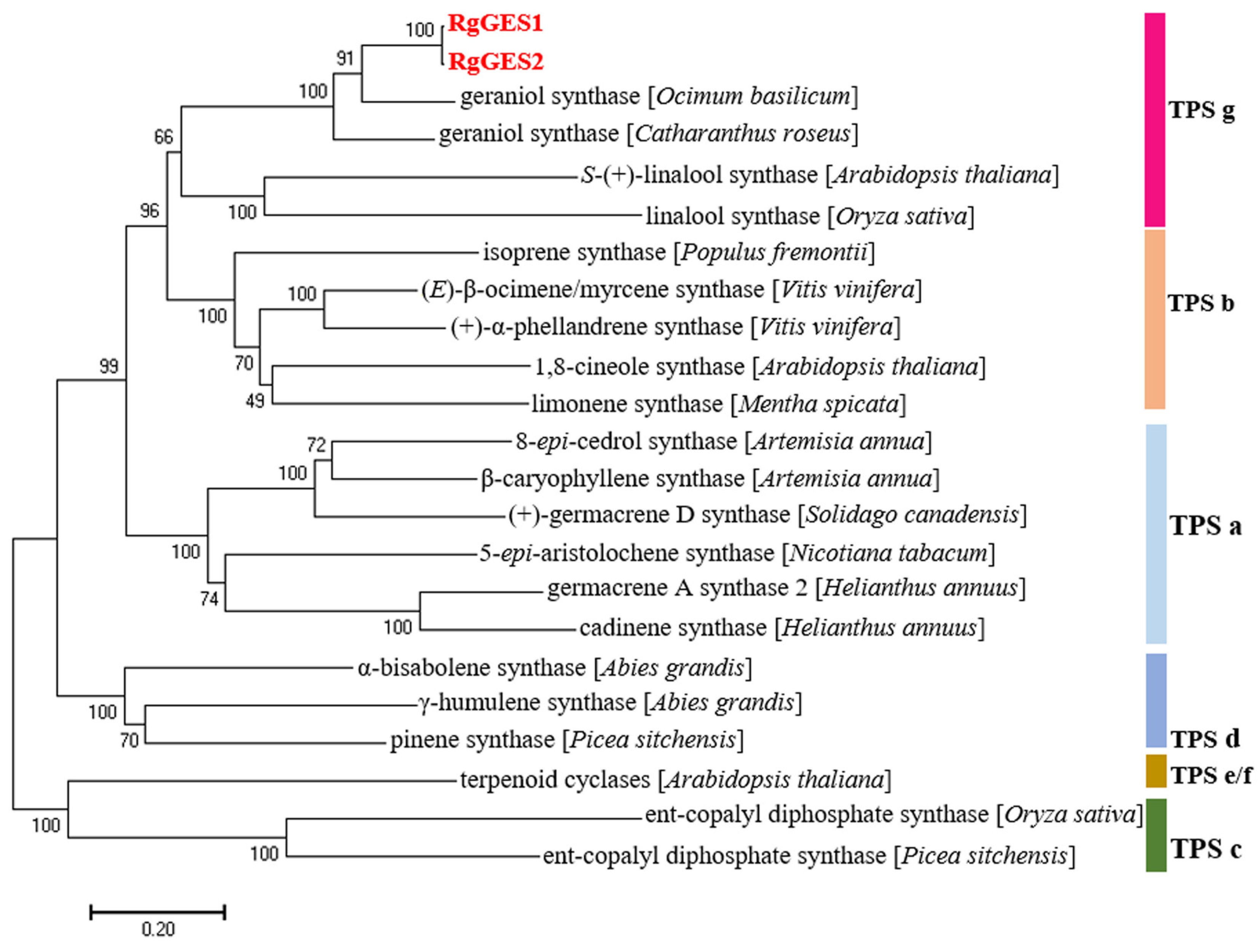
3.5. Cloning and Sequence Analysis of RgGES in R. glutinosa
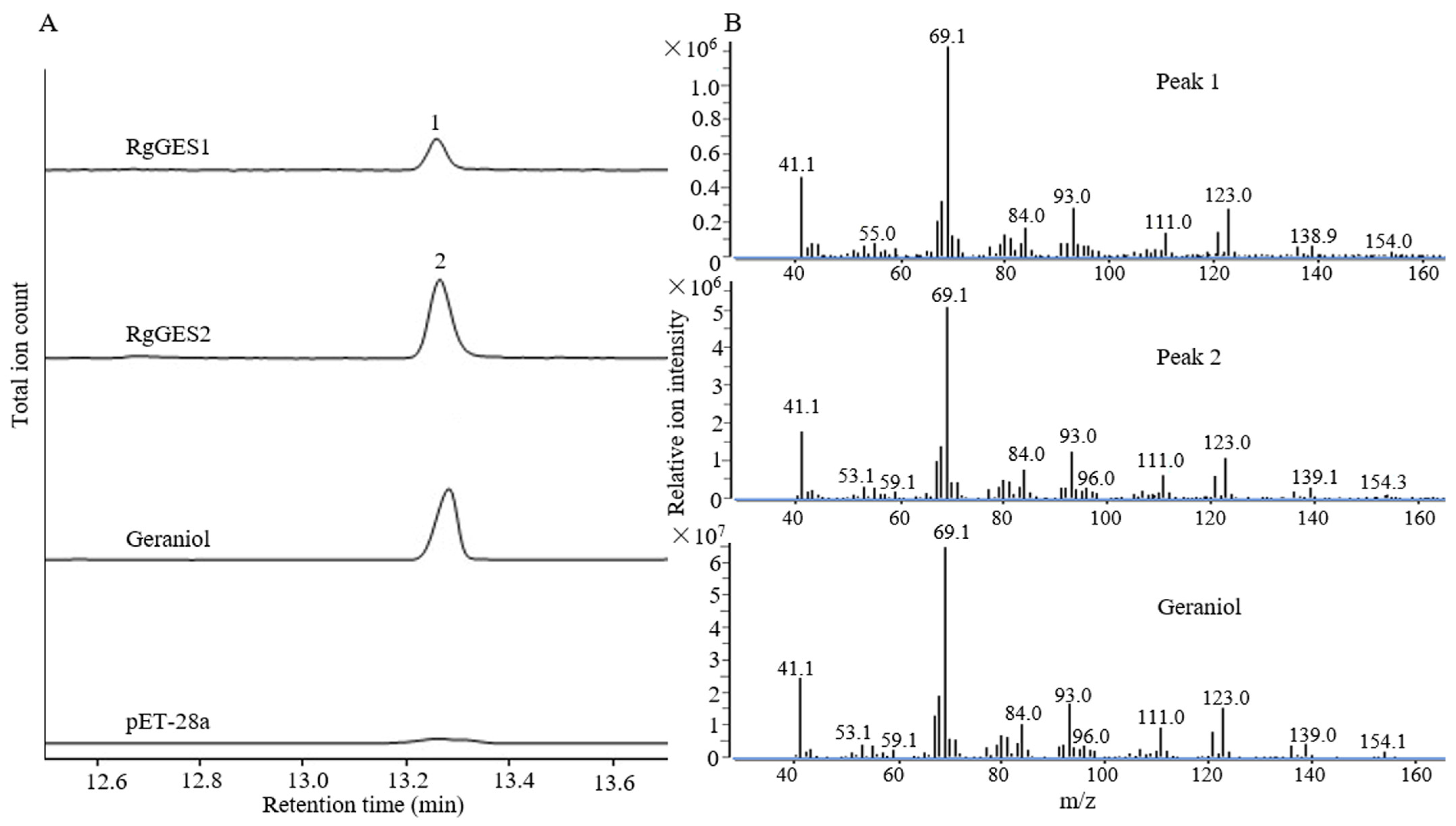
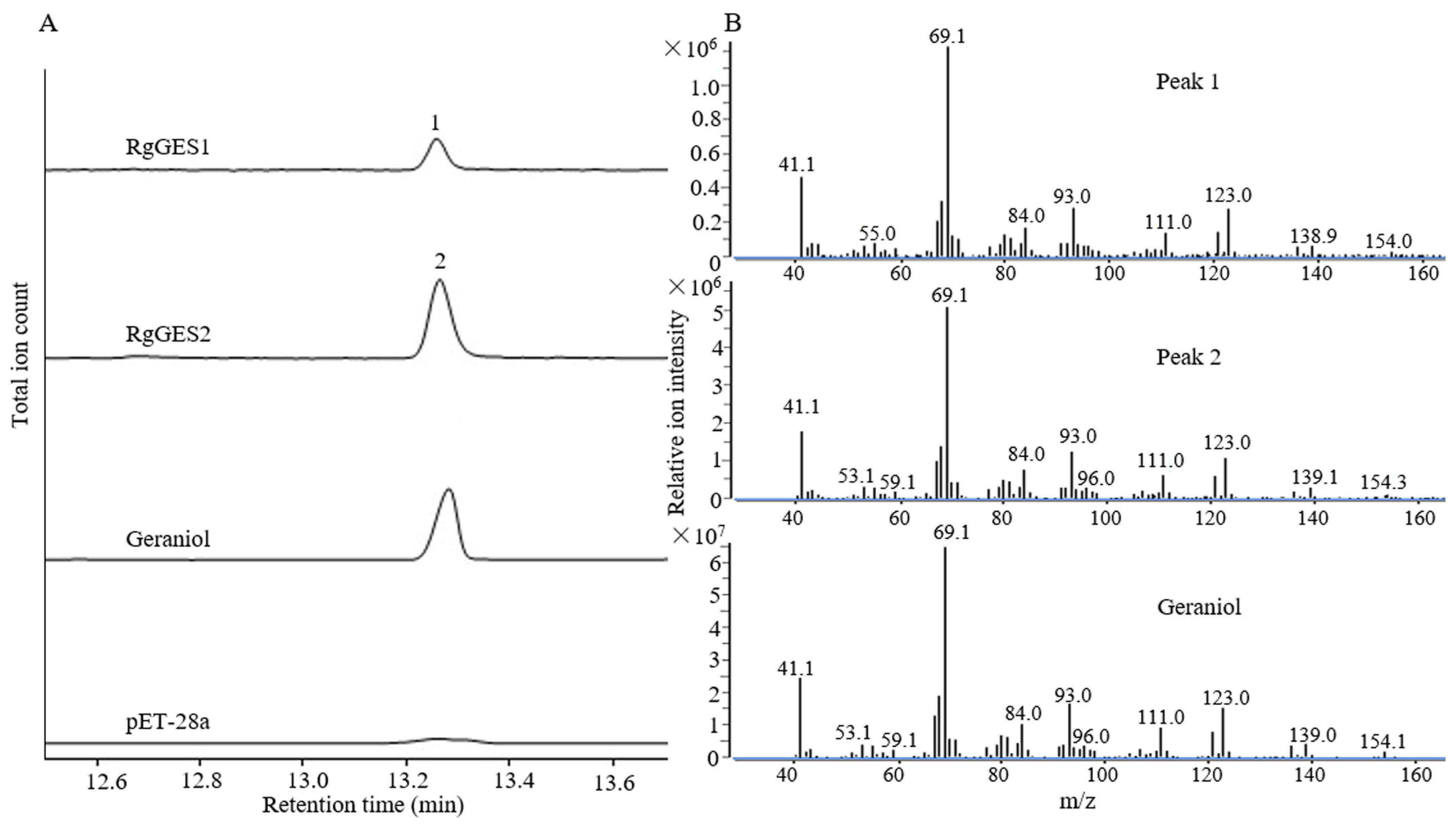
3.6. Functional Characterization of RgGES Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, R.X.; Li, M.X.; Jia, Z.P. Rehmannia glutinosa: Review of botany, chemistry and pharmacology. J. Ethnopharmacol. 2008, 117, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tian, Z.Q.; Xian, X.H.; Yan, C.H.; Li, Q.; Li, N.; Xu, X.K.; Hou, X.J.; Zhang, X.Y.; Yang, Y.N.; et al. Catalpol rescues cognitive deficits by attenuating amyloid β plaques and neuroinflammation. Biomed. Pharmacother. 2023, 165, 115026. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Zhu, P.T.; Zhang, L.; Xiong, B.; Tao, J.H.; Guan, W.; Li, C.L.; Chen, C.; Gu, J.Y.; Duanmu, J.X.; et al. Autophagy inhibition attenuates the induction of anti-inflammatory effect of catalpol in liver fibrosis. Biomed. Pharmacother. 2018, 103, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.T.; Wang, C.Y.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.H.; Liu, K.; Sun, H. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol. Res. 2018, 130, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Yang, L.H.; Zhang, Y.Y.; Niu, C.L.; Cui, Y.; Feng, W.S.; Wang, G.F. BDNF and COX-2 participate in anti-depressive mechanisms of catalpol in rats undergoing chronic unpredictable mild stress. Physiol. Behav. 2015, 151, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Kim, H.J.; Lee, K.H.; Kang, S.C.; Zee, O.P. Antioxidative iridoid glycosides and phenolic compounds from Veronica peregrina. Arch. Pharmacal Res. 2009, 32, 207–213. [Google Scholar] [CrossRef]
- Zhu, P.T.; Wu, Y.; Yang, A.H.; Fu, X.S.; Mao, M.; Liu, Z.G. Catalpol suppressed proliferation, growth and invasion of CT26 colon cancer by inhibiting inflammation and tumor angiogenesis. Biomed. Pharmacother. 2017, 95, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.R. Plant iridoids, their biosynthesis and distribution in angiosperms. In The Proceedings of the Phytochemical Society of Europe; Harborne, J.B., Tomas-Barberan, F.A., Eds.; Clarendon Press: Oxford, UK, 1991; Volume 31, pp. 133–158. [Google Scholar]
- Alagna, F.; Geu-Flores, F.; Kries, H.; Panara, F.; Baldoni, L.; O’Connor, S.E.; Osbourn, A. Identification and characterization of the iridoid synthase involved in Oleuropein biosynthesis in Olive (Olea europaea) Fruits. J. Biol. Chem. 2016, 291, 5542–5554. [Google Scholar] [CrossRef]
- Asada, K.; Salim, V.; Masada-Atsumi, S.; Edmunds, E.; Nagatoshi, M.; Terasaka, K.; Mizukami, H.; De Luca, V. A 7-deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in Madagascar periwinkle. Plant Cell 2013, 25, 4123–4134. [Google Scholar] [CrossRef]
- Collu, G.; Unver, N.; Peltenburg-Looman, A.M.; van der Heijden, R.; Verpoorte, R.; Memelink, J. Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett. 2001, 508, 215–220. [Google Scholar] [CrossRef]
- Geu-Flores, F.; Sherden, N.H.; Courdavault, V.; Burlat, V.; Glenn, W.S.; Wu, C.; Nims, E.; Cui, Y.; O’connor, S.E. An alternative route to cyclic terpenes by reductive cyclization in iridoid biosynthesis. Nature 2012, 492, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Irmler, S.; Schröder, G.; St-Pierre, B.; Crouch, N.P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schroder, J. Indole alkaloid biosynthesis in Catharanthus roseus: New enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.X.; Cong, Y.H.; Zhu, S.L.; Xing, R.L.; Zhang, D.W.; Yao, X.M.; Wan, R.; Wang, Y.; Yu, F. Two classes of cytochrome P450 reductase genes and their divergent functions in Camptotheca acuminata Decne. Int. J. Biol. Macromol. 2019, 138, 1098–1108. [Google Scholar] [CrossRef]
- Krithika, R.; Srivastava, P.L.; Rani, B.; Kolet, S.P.; Chopade, M.; Soniya, M.; Thulasiram, H.V. Characterization of 10-hydroxygeraniol dehydrogenase from Catharanthus roseus reveals cascaded enzymatic activity in iridoid biosynthesis. Sci. Rep. 2015, 5, 8258. [Google Scholar] [CrossRef] [PubMed]
- Salim, V.; Wiens, B.; Masada-Atsumi, S.; Yu, F.; De Luca, V. 7-deoxyloganetic acid synthase catalyzes a key 3 step oxidation to form 7-deoxyloganetic acid in Catharanthus roseus iridoid biosynthesis. Phytochemistry 2014, 101, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Salim, V.; Yu, F.; Altarejos, J.; De Luca, V. Virus-induced gene silencing identifies Catharanthus roseus 7-deoxyloganic acid-7-hydroxylase, a step in iridoid and monoterpene indole alkaloid biosynthesis. Plant J. 2013, 76, 754–765. [Google Scholar] [CrossRef]
- Valletta, A.; Trainotti, L.; Santamaria, A.R.; Pasqua, G. Cell-specific expression of tryptophan decarboxylase and 10-hydroxygeraniol oxidoreductase, key genes involved in camptothecin biosynthesis in Camptotheca acuminata Decne (Nyssaceae). BMC Plant Biol. 2010, 10, 69. [Google Scholar] [CrossRef]
- Xu, C.; Ye, P.; Wu, Q.W.; Liang, S.C.; Wei, W.K.; Yang, J.F.; Chen, W.; Zhan, R.; Ma, D. Identification and functional characterization of three iridoid synthases in Gardenia jasminoides. Planta 2022, 255, 58. [Google Scholar] [CrossRef]
- Damtoft, S.; Jensen, S.R.; Nielsen, B.J.; Thorsen, J. Biosynthesis of iridoid glucosides in Hebenstretia dentata. Phytochemistry 1992, 31, 3839–3843. [Google Scholar] [CrossRef]
- Damtoft, S.; Frederiksen, L.B.; Jensen, S.R. Biosynthesis of iridoid glucosides in Thunbergia alata. Phytochemistry 1994, 37, 1599–1603. [Google Scholar] [CrossRef]
- Sampaio-Santos, M.I.; Kaplan, M.A.C. Biosynthesis significance of iridoids in chemosystematics. J. Braz. Chem. Soc. 2001, 12, 144–153. [Google Scholar] [CrossRef]
- Damtoft, S.; Jensen, S.R.; Jessen, C.U.; Knudsen, T.B. Late stages in the biosynthesis of aucubin in Scrophularia. Phytochemistry 1993, 33, 1089–1093. [Google Scholar] [CrossRef]
- Kumar, V.; Sood, H.; Sharma, M. A proposed biosynthetic pathway of Picrosides linked through the detection of biochemical intermediates in the endangered medicinal herb Picrorhiza kurroa. Phytochem. Anal. 2013, 24, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Kries, H.; Kellner, F.; Kamileen, M.O.; O’Connor, S.E. Inverted stereocontrol of iridoid synthase in snapdragon. J. Biol. Chem. 2017, 292, 14659–14667. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, C.E.; Jiang, Y.D.; Kamileen, M.O.; Lichman, B.R.; Hong, B.K.; Vaillancourt, B.; Buell, C.R.; O’Connor, S.E. Phylogeny-Aware chemoinformatic analysis of chemical diversity in Lamiaceae enables iridoid pathway assembly and discovery of aucubin synthase. Mol. Biol. Evol. 2022, 39, msac057. [Google Scholar] [CrossRef] [PubMed]
- Zhi, J.Z.; Li, Y.J.; Zhang, Z.Y.; Yang, C.F.; Geng, X.T.; Zhang, M.; Li, X.R.; Zuo, X.; Li, M.J.; Huang, Y.; et al. Molecular regulation of catalpol and acteoside accumulation in radial striation and non-radial striation of Rehmannia glutinosa tuberous root. Int. J. Mol. Sci. 2018, 19, 3751. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.G.; Dong, C.M.; Song, C.; Wang, X.L.; Zheng, X.K.; Niu, Y.; Chen, S.; Feng, W. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 2021, 19, 3954–3963. [Google Scholar] [CrossRef]
- Kumar, V.; Chauhan, R.S.; Tandon, C. Biosynthesis and therapeutic implications of iridoid glycosides from Picrorhiza genus: The road ahead. J. Plant Biochem. Biotechnol. 2017, 26, 1–13. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G. Automated genome annotation and pathway identification using the KEGG orthology as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Chen, Q.J.; Li, X.E. Selection of the reference genes for gene expression studies in Rehmannia glutinosa by real-time quantitative PCR. Chin. Agric. Sci. Bull. 2011, 27, 76–82. [Google Scholar]
- Zhao, C.H.; Yu, Z.M.; da Silva, J.A.T.; He, C.M.; Wang, H.B.; Si, C.; Zhang, M.; Zeng, D.; Duan, J. Functional characterization of a Dendrobium officinale geraniol synthase DoGES1 involved in floral scent formation. Int. J. Mol. Sci. 2020, 21, 7005. [Google Scholar] [CrossRef] [PubMed]
- Simkin, A.J.; Miettinen, K.; Claudel, P.; Burlat, V.; Guirimand, G.; Courdavault, V.; Papon, N.; Meyer, S.; Godet, S.; St-Pierre, B.; et al. Characterization of the plastidial geraniol synthase from Madagascar periwinkle which initiates the monoterpenoid branch of the alkaloid pathway in internal phloem. Phytochemistry 2013, 85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Podust, L.M.; Sherman, D.H. Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep. 2012, 29, 1251–1266. [Google Scholar] [CrossRef]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Degenhardt, J.; Kollner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.F.; Liu, J.; Zhang, J.; Liu, F.G.; Wei, B. Content comparison of catalpol and acteoside in Rehmannia glutinosa with different varieties from genuine producing area. ZhongguoShiyanFangjixueZazhi 2013, 19, 97–101. [Google Scholar]
- Ji, X.Q.; Sun, P.; Qi, J.J.; Liao, D.Q.; Li, X.E. Study on distribution and dynamic accumulation of catalpol and total iridoid in fresh Rehmannia glutinosa. Zhongguo Zhongyao Zazhi 2014, 39, 466–470. [Google Scholar] [PubMed]
- Li, M.J.; Yang, Y.H.; Chen, X.J.; Wang, F.Q.; Lin, W.X.; Yi, Y.J.; Zeng, L.; Yang, S.Y.; Zhang, Z.Y. Transcriptome/Degradome-wide identification of R. glutinosa miRNAs and their targets: The role of miRNA activity in the replanting disease. PLoS ONE 2013, 8, e68531. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xiao, X.G.; Duan, L.S.; Guo, Y.H.; Qi, J.J.; Liao, D.Q.; Zhao, C.; Liu, Y.; Zhou, L.; Li, X. Dynamic transcriptional profiling provides insights into tuberous root development in Rehmannia glutinosa. Front. Plant Sci. 2015, 6, 396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Q.; Wang, X.N.; Wang, W.S.; Duan, H.Y. De novo transcriptome sequencing-based discovery and expression analyses of verbascoside biosynthesis-associated genes in Rehmannia glutinosa tuberous roots. Mol. Breed. 2016, 36, 139. [Google Scholar] [CrossRef]
- Wang, F.Q.; Zhi, J.Y.; Zhang, Z.Y.; Wang, L.N.; Suo, Y.F.; Xie, C.X.; Li, M.J.; Zhang, B.; Du, J.F.; Gu, L.; et al. Transcriptome analysis of salicylic acid treatment in Rehmannia glutinosa hairy roots using RNA-seq technique for identification of genes involved in acteoside biosynthesis. Front. Plant Sci. 2017, 8, 787. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Song, S.H.; Zhou, L.L.; Zhang, B.; Qi, J.J.; Li, X.N. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int. J. Mol. Sci. 2012, 13, 13748–13763. [Google Scholar] [CrossRef]
- Zhou, P.F.; Li, H.H.; Lin, Y.J.; Zhou, Y.J.; Chen, Y.Z.; Li, Y.H.; Li, X.; Yan, H.; Lin, W.M.; Xu, B.L.; et al. Omics analyses of Rehmannia glutinosa dedifferentiated and cambial meristematic cells reveal mechanisms of catalpol and indole alkaloid biosynthesis. BMC Plant Biol. 2023, 23, 463. [Google Scholar] [CrossRef]
- Dudareva, N.; Martin, D.; Kish, C.M.; Kolosova, N.; Gorenstein, N.; Faldt, J.; Miller, B.; Bohlmann, J. (E)-β-ocimene and myrcene synthase genes of floral scent biosynthesis in snapdragon: Function and expression of three terpene synthase genes of a new terpene synthase subfamily. Plant Cell 2003, 15, 1227–1241. [Google Scholar] [CrossRef]
- Ye, P.; Liang, S.C.; Wang, X.M.; Duan, L.X.; Jiang, F.Y.Y.; Yang, J.F.; Zhan, R.; Ma, D. Transcriptome analysis and targeted metabolic profiling for pathway elucidation and identification of a geraniol synthase involved in iridoid biosynthesis from Gardenia jasminoides. Ind. Crops Prod. 2019, 132, 48–58. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error Rate | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| J9L | 31,180,906 | 29,195,338 | 8.76 G | 0.03 | 97.84 | 93.43 | 45.42 |
| J9R | 31,674,784 | 30,297,505 | 9.09 G | 0.03 | 97.85 | 93.38 | 44.44 |
| J9AR | 30,827,652 | 29,371,270 | 8.81 G | 0.03 | 97.75 | 93.19 | 45.10 |
| BJL | 30,882,434 | 29,153,263 | 8.75 G | 0.03 | 97.83 | 93.36 | 45.02 |
| HFL | 31,220,770 | 29,902,124 | 8.97 G | 0.03 | 98.01 | 93.91 | 44.47 |
| Databases | Number of Unigenes | Percentage |
|---|---|---|
| Annotated in all databases | 6958 | 9.78 |
| Annotated in KOG | 10,119 | 14.22 |
| Annotated in KEGG | 13,862 | 19.48 |
| Annotated in GO | 28,511 | 40.07 |
| Annotated in Pfam | 28,512 | 40.07 |
| Annotated in Swiss-Prot | 29,731 | 41.79 |
| Annotated in Nt | 30,861 | 43.37 |
| Annotated in Nr | 38,788 | 54.52 |
| Annotated in at least one database | 71,142 | 100 |
| Total unigenes | 71,142 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhai, X.; Ma, L.; Zhao, L.; An, N.; Feng, W.; Huang, L.; Zheng, X. Transcriptome Analysis Provides Insights into Catalpol Biosynthesis in the Medicinal Plant Rehmannia glutinosa and the Functional Characterization of RgGES Genes. Genes 2024, 15, 155. https://doi.org/10.3390/genes15020155
Li Y, Zhai X, Ma L, Zhao L, An N, Feng W, Huang L, Zheng X. Transcriptome Analysis Provides Insights into Catalpol Biosynthesis in the Medicinal Plant Rehmannia glutinosa and the Functional Characterization of RgGES Genes. Genes. 2024; 15(2):155. https://doi.org/10.3390/genes15020155
Chicago/Turabian StyleLi, Yuanjun, Xiaoru Zhai, Ligang Ma, Le Zhao, Na An, Weisheng Feng, Longyu Huang, and Xiaoke Zheng. 2024. "Transcriptome Analysis Provides Insights into Catalpol Biosynthesis in the Medicinal Plant Rehmannia glutinosa and the Functional Characterization of RgGES Genes" Genes 15, no. 2: 155. https://doi.org/10.3390/genes15020155
APA StyleLi, Y., Zhai, X., Ma, L., Zhao, L., An, N., Feng, W., Huang, L., & Zheng, X. (2024). Transcriptome Analysis Provides Insights into Catalpol Biosynthesis in the Medicinal Plant Rehmannia glutinosa and the Functional Characterization of RgGES Genes. Genes, 15(2), 155. https://doi.org/10.3390/genes15020155