


Identification of Retrocopies in Lepidoptera and Impact on Domestication of Silkworm


Abstract

1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Identifying Retrocopies in Lepidopteran Insect
2.3. Sequence Features, Divergence Times, Potential Functions, and Orthology of Retrocopies Within Lepidopteran Insect Genomes
2.4. Analysis of the Potential Function of Retrocopy in B. mori
2.5. RT-PCR
3. Results
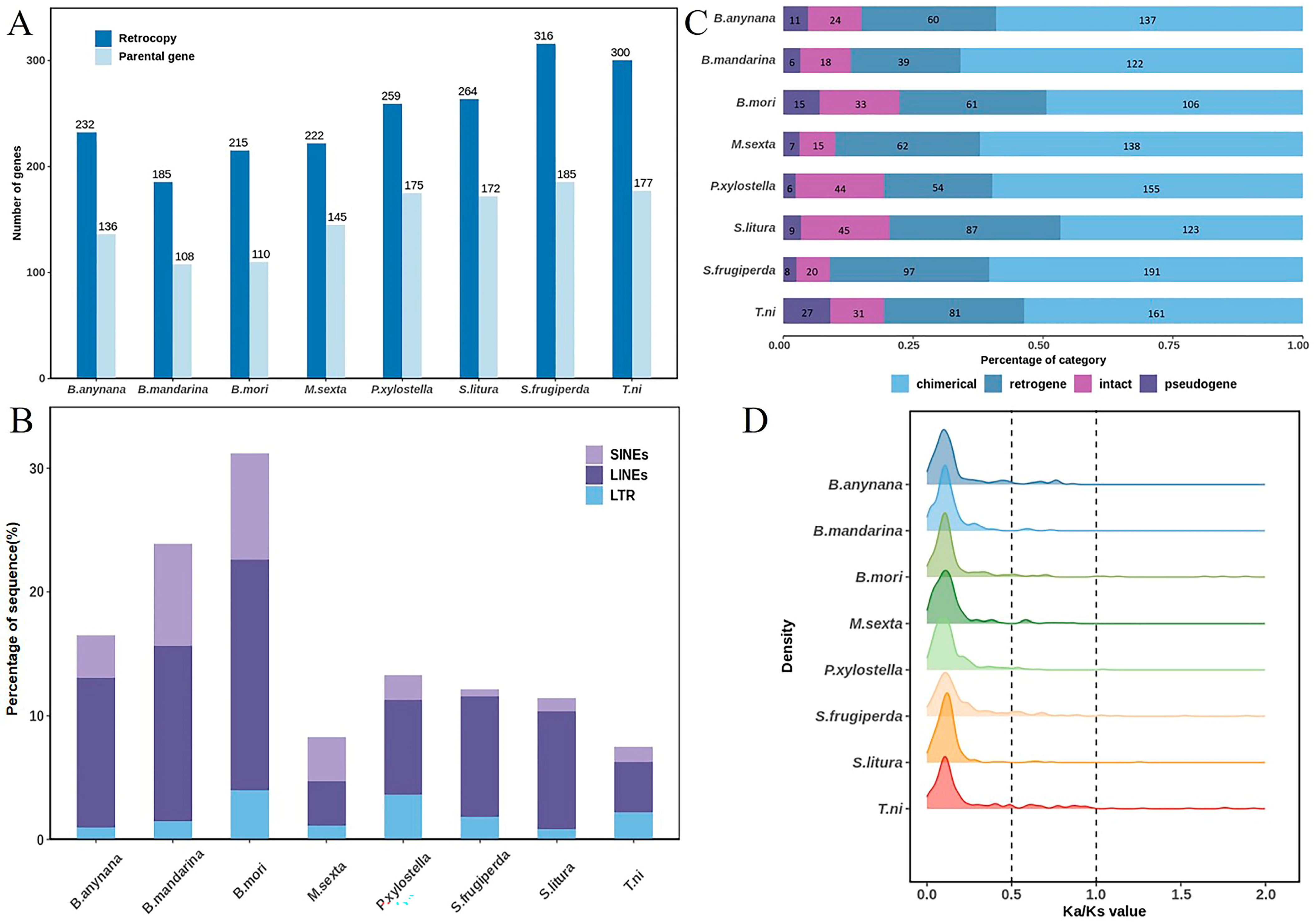
3.1. Identification of Lepidopteran Retrocopies
3.2. Expression and Functional Analysis of Lepidopteran Retrocopies
3.3. Age Distribution and Evolutionary Analysis of Lepidopteran Retrocopies
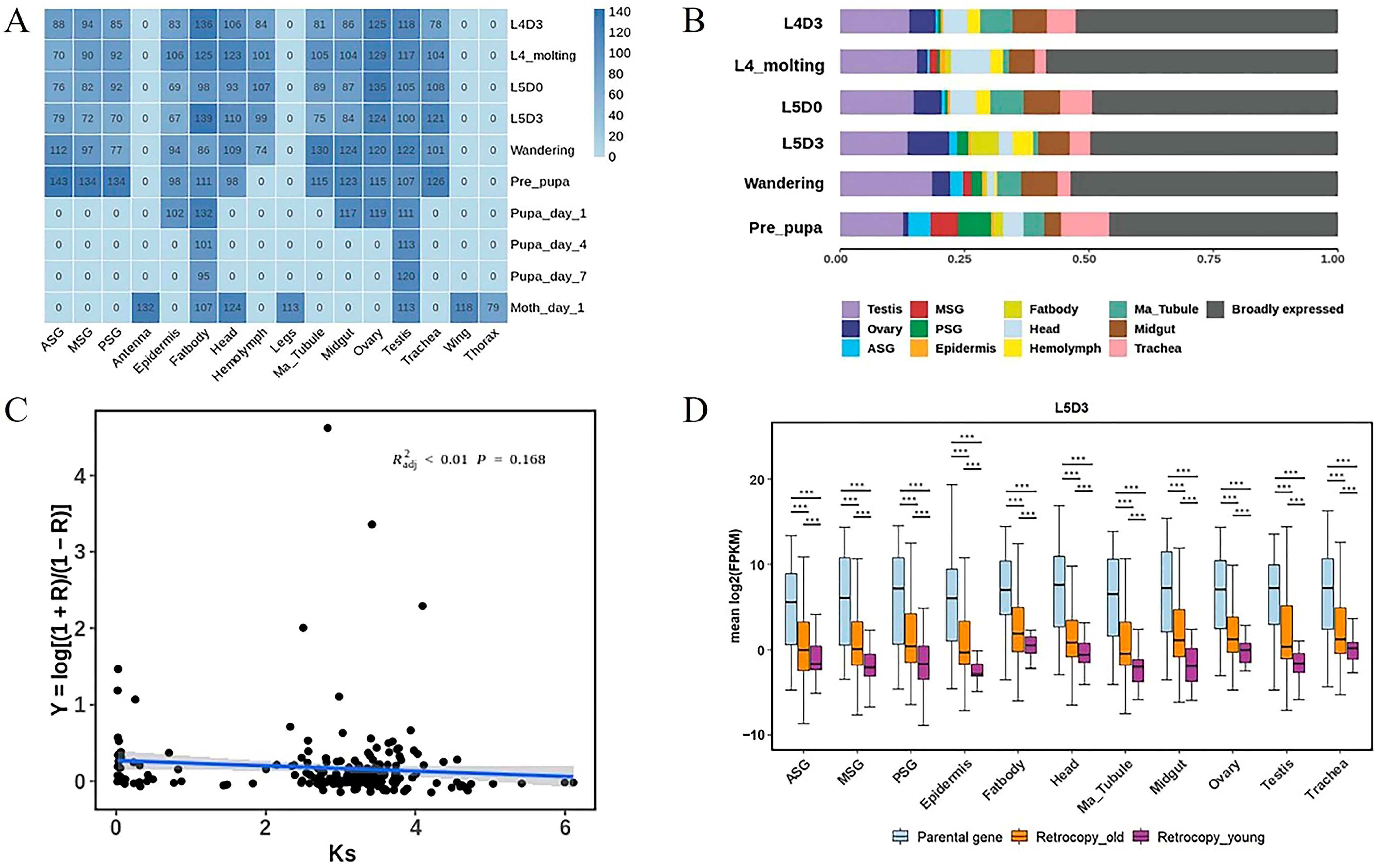
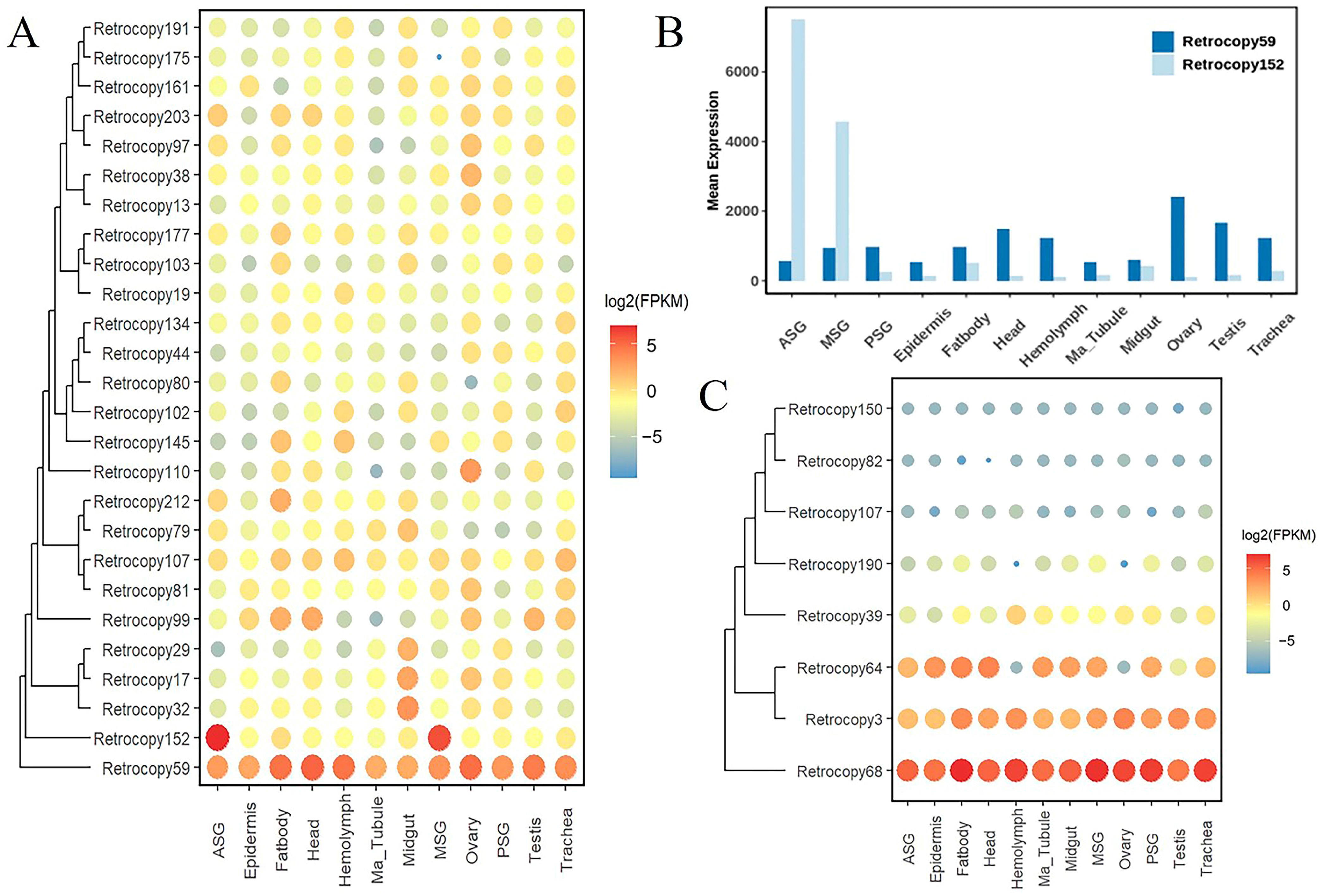
3.4. Transcriptome and Functional Analysis of Silkworm Retrocopies
3.5. Impact of Retrocopies on Silkworm Domestication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Triant, D.A.; Cinel, S.D.; Kawahara, A.Y. Lepidoptera genomes: Current knowledge, gaps and future directions. Curr. Opin. Insect Sci. 2018, 25, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A. Core questions in domestication research. Proc. Natl. Acad. Sci. USA 2015, 112, 3191–3198. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Hattori, M.; Yada, T.; Gojobori, T.; Sakaki, Y.; Okada, N. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003, 4, R74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Harrison, P.M.; Liu, Y.; Gerstein, M. Millions of years of evolution preserved: A comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003, 13, 2541–2558. [Google Scholar] [CrossRef]
- Vinckenbosch, N.; Dupanloup, I.; Kaessmann, H. Evolutionary fate of retroposed gene copies in the human genome. Proc. Natl. Acad. Sci. USA 2006, 103, 3220–3225. [Google Scholar] [CrossRef]
- Du, K.; He, S. Evolutionary fate and implications of retrocopies in the African coelacanth genome. BMC Genom. 2015, 16, 915. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Yang, L.; Zhang, Y.E.; Xue, Y.; He, S. Correlated expression of retrocopies and parental genes in zebrafish. Mol. Genet. Genom. 2016, 291, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Y.; Liu, Y.; Han, B. Computational identification of 69 retroposons in Arabidopsis. Plant Physiol. 2005, 138, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Mizuno, H.; Kawahara, Y.; Wakimoto, H.; Ikawa, H.; Kawahigashi, H.; Kanamori, H.; Matsumoto, T.; Itoh, T.; Gaut, B.S. Retrogenes in rice (Oryza sativa L. ssp. japonica) exhibit correlated expression with their source genes. Genome Biol. Evol. 2011, 3, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, Y.; Long, M. Extensive structural renovation of retrogenes in the evolution of the Populus genome. Plant Physiol. 2009, 151, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Jakalski, M.; Takeshita, K.; Deblieck, M.; Koyanagi, K.O.; Makalowska, I.; Watanabe, H.; Makalowski, W. Comparative genomic analysis of retrogene repertoire in two green algae Volvox carteri and Chlamydomonas reinhardtii. Biol. Direct 2016, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.S.; Casola, C.; Feschotte, C.; Betrán, E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007, 8, R11. [Google Scholar] [CrossRef]
- Schrider, D.R.; Stevens, K.; Cardeño, C.M.; Langley, C.H.; Hahn, M.W. Genome-wide analysis of retrogene polymorphisms in Drosophila melanogaster. Genome Res. 2011, 21, 2087–2095. [Google Scholar] [CrossRef]
- Xu, P.; Feuda, R.; Lu, B.; Xiao, H.; Graham, R.I.; Wu, K. Functional opsin retrogene in nocturnal moth. Mob. DNA 2016, 7, 18. [Google Scholar] [CrossRef]
- Toups, M.A.; Pease, J.B.; Hahn, M.W. No excess gene movement is detected off the avian or lepidopteran Z chromosome. Genome Biol. Evol. 2011, 3, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Long, M.; Vibranovski, M.D. Retrogenes moved out of the z chromosome in the silkworm. J. Mol. Evol. 2012, 74, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Tellegen, A.R.; Dessing, A.J.; Houben, K.; Riemers, F.M.; Creemers, L.B.; Mastbergen, S.C.; Meij, B.P.; Miranda-Bedate, A.; Tryfonidou, M.A. Dog as a Model for Osteoarthritis: The FGF4 Retrogene Insertion May Matter. J. Orthop. Res. 2019, 37, 2550–2560. [Google Scholar] [CrossRef]
- Parker, H.G.; VonHoldt, B.M.; Quignon, P.; Margulies, E.H.; Shao, S.; Mosher, D.S.; Spady, T.C.; Elkahloun, A.; Cargill, M.; Jones, P.G.; et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science 2009, 325, 995–998. [Google Scholar] [CrossRef]
- Batcher, K.; Dickinson, P.; Maciejczyk, K.; Brzeski, K.; Rasouliha, S.H.; Letko, A.; Drogemuller, C.; Leeb, T.; Bannasch, D. Multiple FGF4 Retrocopies Recently Derived within Canids. Genes 2020, 11, 839. [Google Scholar] [CrossRef]
- Lee, Y.C.G.; Ventura, I.M.; Rice, G.R.; Chen, D.Y.; Colmenares, S.U.; Long, M. Rapid Evolution of Gained Essential Developmental Functions of a Young Gene via Interactions with Other Essential Genes. Mol. Biol. Evol. 2019, 36, 2212–2226. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Z.; Vizcay-Barrena, G.; Shi, J.; Liang, W.; Yuan, Z.; Werck-Reichhart, D.; Schreiber, L.; Wilson, Z.A.; Zhang, D. ABORTED MICROSPORES Acts as a Master Regulator of Pollen Wall Formation in Arabidopsis. Plant Cell 2014, 26, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 4, 4–10. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating γ-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Krinsky, B.H.; Long, M. New genes as drivers of phenotypic evolution. Nat. Rev. Genet. 2013, 14, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Betran, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef]
- Gao, X.; Li, Y.; Adetula, A.A.; Wu, Y.; Chen, H. Analysis of new retrogenes provides insight into dog adaptive evolution. Ecol. Evol. 2019, 9, 11185–11197. [Google Scholar] [CrossRef]
- Betran, E.; Thornton, K.; Long, M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002, 12, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Mathias, S.L.; Scott, A.F.; Kazazian, H.H., Jr.; Boeke, J.D.; Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science 1991, 254, 1808–1810. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.D.; Ling, H.Q. Non-LTR retrotransposons: LINEs and SINEs in plant genome. Yi Chuan 2006, 28, 731–736. [Google Scholar]
- Fu, B.D.; Chen, M.; Zou, M.; Long, M.Y.; He, S.P. The rapid generation of chimerical genes expanding protein diversity in zebrafish. Bmc Genom. 2010, 11, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zou, M.; Duan, M.; He, S.; Wang, G. Identification and analysis of retrogenes in the East Asian nematode Caenorhabditis sp. 5 genome. Genome 2015, 58, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.J.; Kaessmann, H.; Betran, E.; Long, M. Extensive gene traffic on the mammalian X chromosome. Science 2004, 303, 537–540. [Google Scholar] [CrossRef]
- Carelli, F.N.; Hayakawa, T.; Go, Y.; Imai, H.; Warnefors, M.; Kaessmann, H. The life history of retrocopies illuminates the evolution of new mammalian genes. Genome Res. 2016, 26, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.C.; Galante, P.A. A Genome-Wide Landscape of Retrocopies in Primate Genomes. Genome Biol. Evol. 2015, 7, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.Y.; Zhou, Z.Y.; Lu, C.; Cheng, D.J.; Dai, F.Y.; Li, B.; Zhao, P.; Zha, X.F.; Cheng, T.C.; Chai, C.L.; et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 2004, 306, 1937–1940. [Google Scholar]
- Marques, A.C.; Dupanloup, I.; Vinckenbosch, N.; Reymond, A.; Kaessmann, H. Emergence of young human genes after a burst of retroposition in primates. PLoS Biol. 2005, 3, e357. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Guo, Y.; Zhang, Z.; Li, D.; Xuan, Z.; Li, Z.; Dai, F.; Li, Y.; Cheng, D.; Li, R.; et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 2009, 326, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.Y.; Wang, J.; Zhou, Z.Y.; Li, R.Q.; Fan, W.; Cheng, D.J.; Cheng, T.C.; Qin, J.J.; Duan, J.; Xu, H.F.; et al. The genome of a lepidopteran model insect, the silkworm. Insect Biochem. Mol. 2008, 38, 1036–1045. [Google Scholar]
- Guerreiro, M.P.G. What makes transposable elements move in the genome? Heredity 2012, 108, 461–468. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Movement | Expected | Observed | Excess (%) |
|---|---|---|---|
| Z→A | 7 | 6 | −14.28 |
| A→Z | 8 | 12 | 50 |
| A→A | 200 | 197 | −1.5 |
| Gene | Ka/Ks < 0.5 | Ka/Ks = 0.5–1.0 | Ka/Ks > 1.0 | Mean Ka/Ks |
|---|---|---|---|---|
| Intact retrocopies | 1761 (92.5%) | 96 (5.04%) | 33 (1.7%) | 0.4666 |
| Retropseudogenes | 66 (74.2%) | 18 (20.2%) | 5 (5.6%) | 0.9146 |
| Evolutionary Period (Ma) | Branch Number | Number of Retrocopies | Evolutionary Period (Myr) | Average of Retrocopy (Myr) |
|---|---|---|---|---|
| 114–156 | 8 | 14 | 42 | 0.333 |
| 111–114 | 7 | 2 | 3 | 0.667 |
| 67–111 | 6 | 13 | 44 | 0.295 |
| 60–111 | 5 | 34 | 51 | 0.667 |
| 16.9–60 | 4 | 73 | 43.1 | 1.691 |
| 0.0051–67 | 3 | 81 | 66.99 | 1.209 |
| Retrocopy | Parental Gene | Host Gene | Gene Type | Functional |
|---|---|---|---|---|
| Retrocopy3 | Chr15.332 | Chr01.171 | Retrogene | ubiquitin-protein transferase activity |
| Retrocopy39 | Chr15.790 | Chr05.600 | Chimerical | peptidyl-prolyl cis-trans isomerase |
| Retrocopy64 | Chr24.497 | Chr09.305 | Retrogene | protein binding |
| Retrocopy68 | Chr19.416 | Chr09.504 | Chimerical | peptidyl-prolyl cis-trans isomerase |
| Retrocopy82 | Chr01.34 | Chr11.621 | Chimerical | involved in glutamine metabolism |
| Retrocopy107 | Chr07.550 | Chr15.738 | Intact retrocopy | uncharacterized protein |
| Retrocopy150 | Chr22.674 | NA | Retropseudogene | uncharacterized protein |
| Retrocopy190 | Chr11.228 | Chr25.579 | Chimerical | oxidoreductase activity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bie, L.; Sun, J.; Wang, Y.; Wang, C. Identification of Retrocopies in Lepidoptera and Impact on Domestication of Silkworm. Genes 2024, 15, 1641. https://doi.org/10.3390/genes15121641
Bie L, Sun J, Wang Y, Wang C. Identification of Retrocopies in Lepidoptera and Impact on Domestication of Silkworm. Genes. 2024; 15(12):1641. https://doi.org/10.3390/genes15121641
Chicago/Turabian StyleBie, Lingzi, Jiahe Sun, Yi Wang, and Chunfang Wang. 2024. "Identification of Retrocopies in Lepidoptera and Impact on Domestication of Silkworm" Genes 15, no. 12: 1641. https://doi.org/10.3390/genes15121641
APA StyleBie, L., Sun, J., Wang, Y., & Wang, C. (2024). Identification of Retrocopies in Lepidoptera and Impact on Domestication of Silkworm. Genes, 15(12), 1641. https://doi.org/10.3390/genes15121641