
Mitochondrial Genome Assembly and Comparative Analysis of Chionanthus Retusus (Oleaceae)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Genome Sequencing
2.2. Genome Assembly and Annotation
2.3. Analysis of RSCU and RNA Editing Sites Prediction
2.4. Analysis of Sequence Repeats
2.5. Chloroplast to Mitochondrion DNA Transformation
2.6. Synteny and Phylogenetic Analysis
3. Results
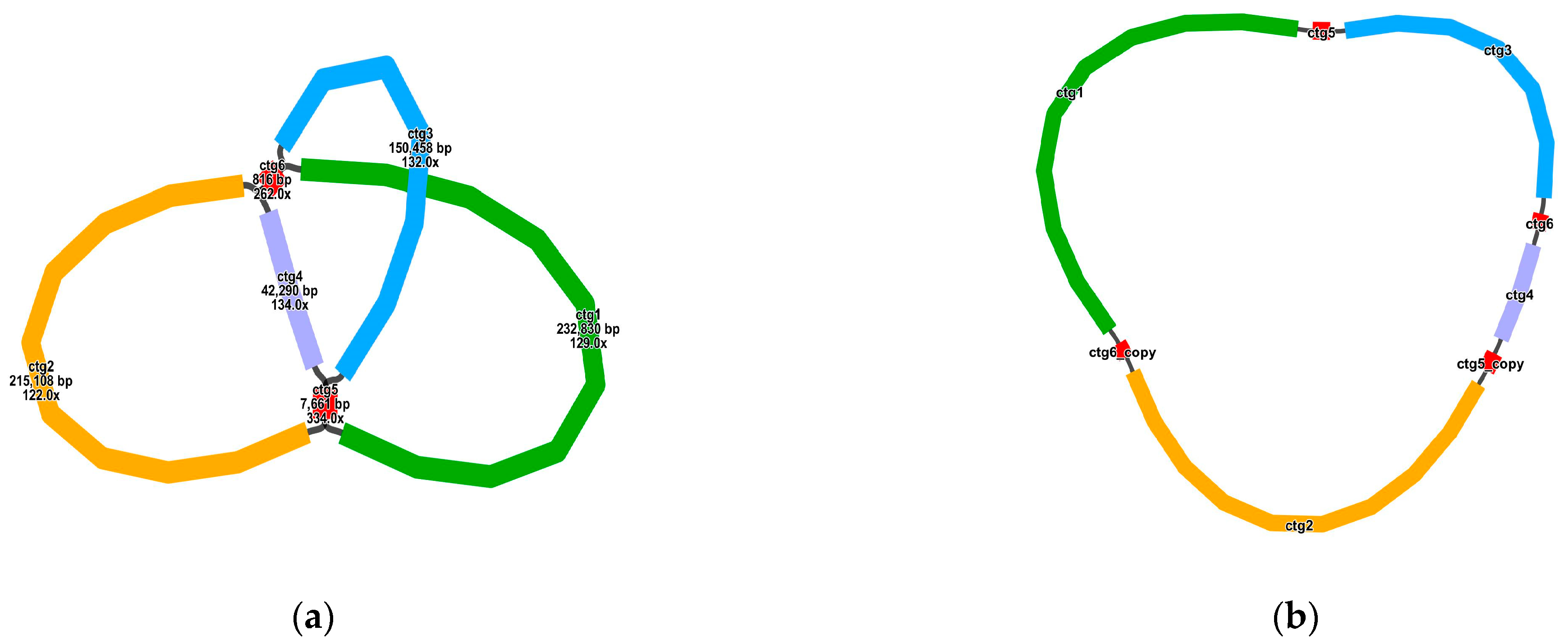
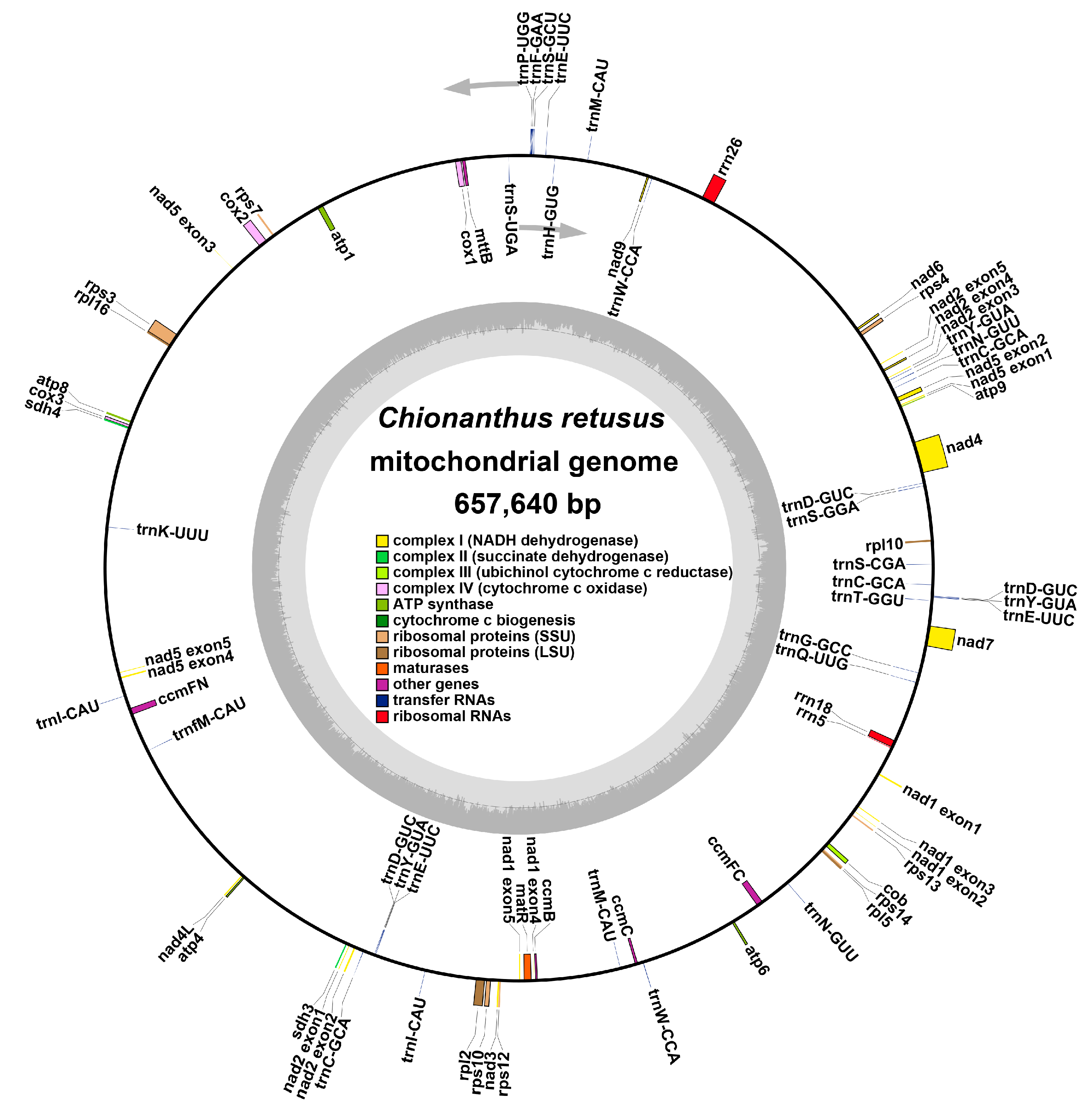
3.1. C. Retusus Mitochondrial Genome Characteristics
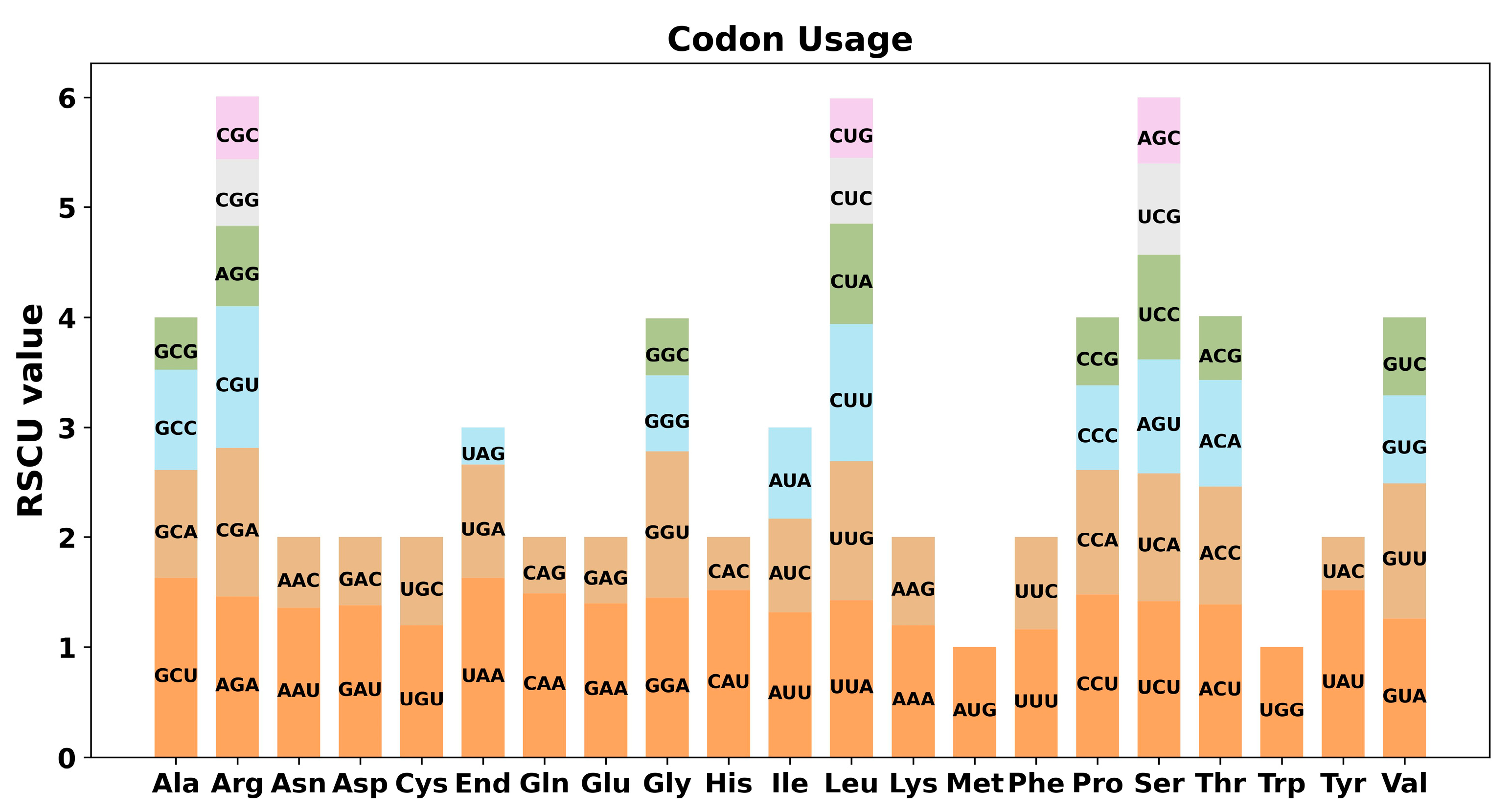
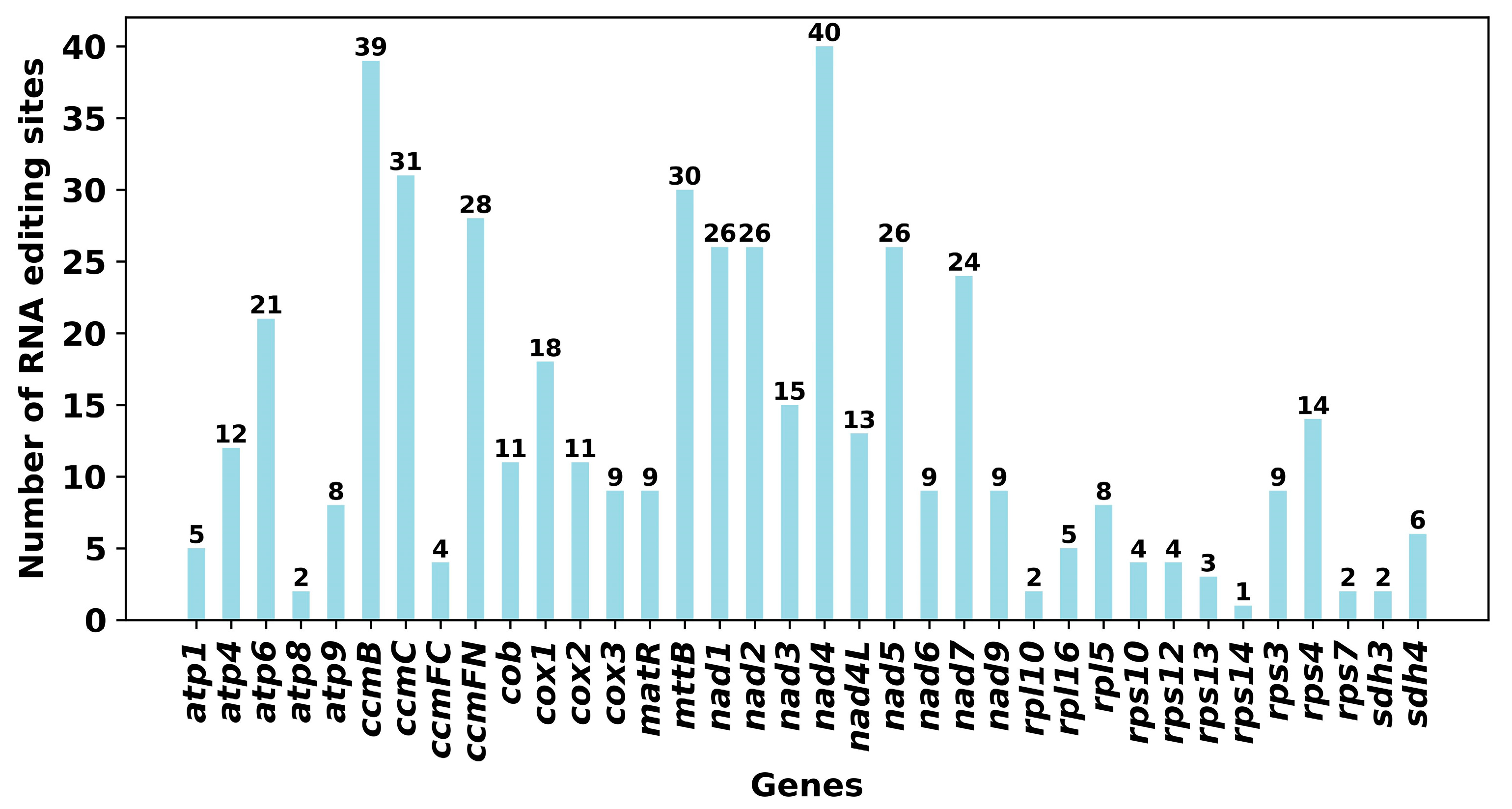
3.2. Codon Usage Preference and RNA Editing Sites Prediction
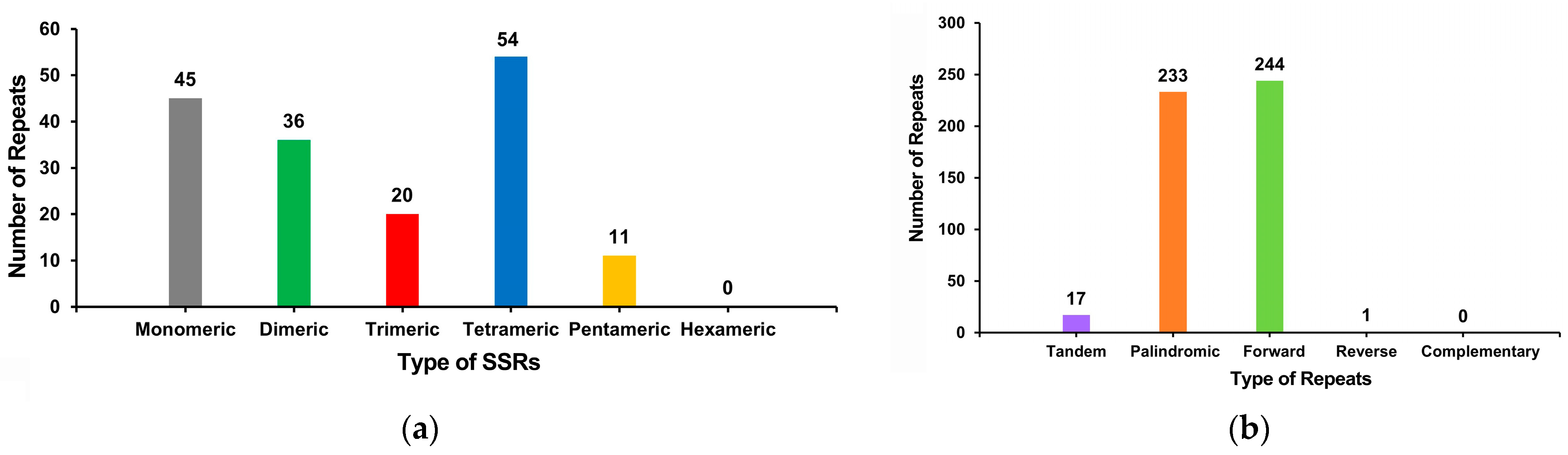
3.3. C. Retusus Mitochondrial Genome Repeat Sequence Analysis
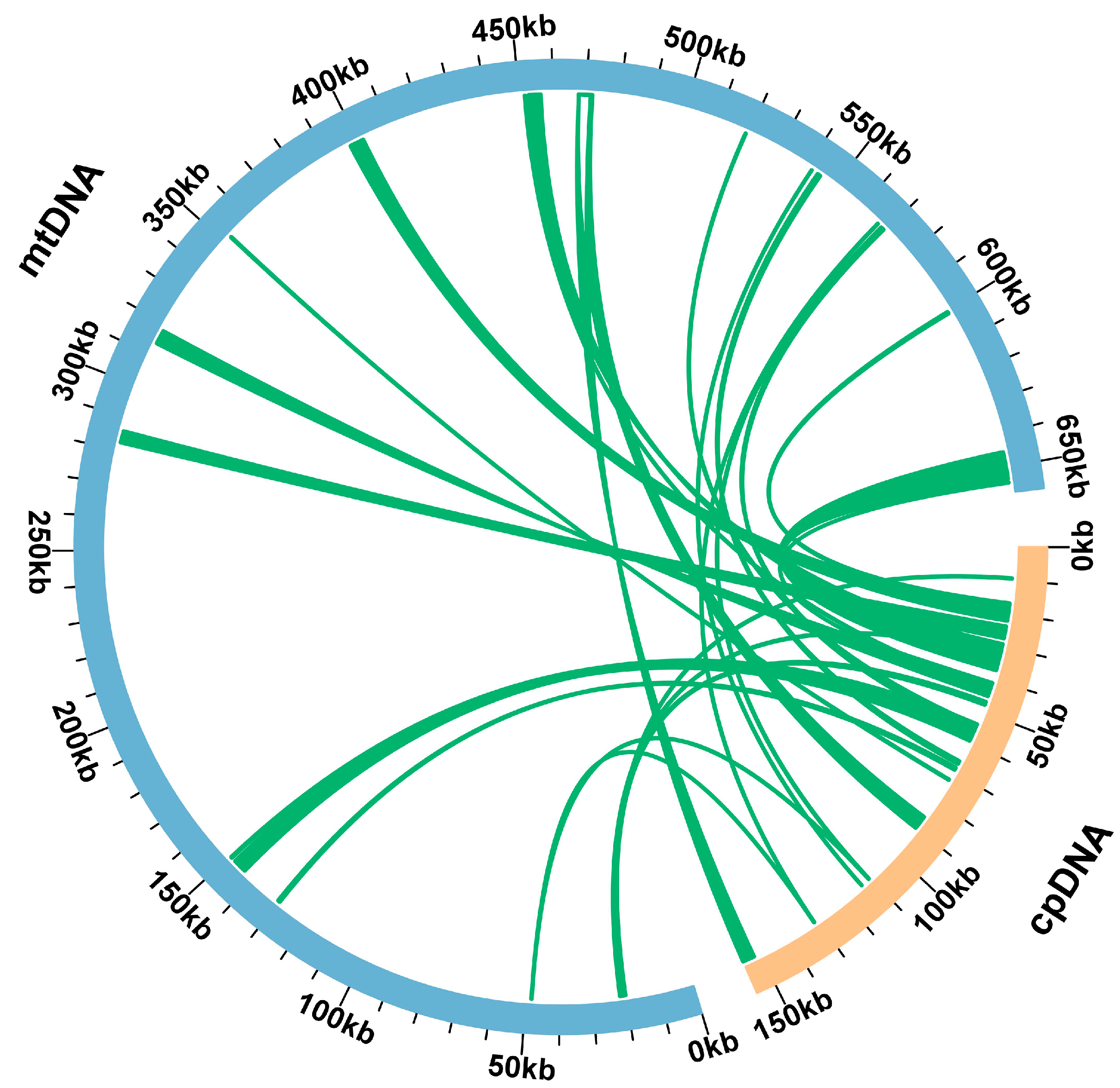
3.4. Chloroplast to Mitochondrion DNA Transformation of C. retusus
3.5. Synteny and Phylogenetic Analysis of C. retusus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, T.R. Growth and Photosynthetic Physiological Characteristics Responseof Chionanthus retusus Seedlings to Water and Salt Stress. Master’s Thesis, Shandong Agricultural University, Taian, China, 2021. [Google Scholar]
- Wang, Q.J.; Liu, J. Yuanlin Shumu, 1st ed.; China Agricultural University Press: Beijing, China, 2017; p. 134. [Google Scholar]
- Qu, K. Collection and Evaluation of Chionanthus retusus Germplasm Resources and Analysis of Genetic Diversity. Master’s Thesis, Shandong Agricultural University, Taian, China, 2019. [Google Scholar]
- Fang, L. The comprehensive utilization value and cultivation management technology of Chionanthus retusus. Mod. Agric. Sci. Technol. 2017, 123–124. [Google Scholar]
- Bai, Z.H. Cuttage propagation technique of hard branch in small bow-shed hotbed of Chionanthus retusus. For. Sci. Technol. 2007, 39–40. [Google Scholar] [CrossRef]
- Yu, F.L. Research on grafting propagation technology of Chionanthus retusus seedlings. For. Sci. Technol. 2007, 27–28. [Google Scholar] [CrossRef]
- Wang, X.M. Propagation and cultivation of Chionanthus retusus. For. Sci. Technol. 1999, 42. [Google Scholar] [CrossRef]
- Hu, X.L.; Jiang, Q.; Yin, F.J.; Liu, T. Extraction and Determination of Mineral Elements and Bioactive Components in Chinese Fringe Tree Young Leaf Tea and Flower Tea. J. Food Sci. 2010, 31, 112–115. [Google Scholar]
- Deng, R.X.; Lu, Z.Y.; Zhang, C.F.; Duan, W.L.; Yin, W.P. Chemical Constituents from Flowers of Chionanthus retus. J. Henan Univ. Sci. Technol. (Nat. Sci.) 2013, 34, 92–95+9. [Google Scholar]
- Zhang, J.F.; Li, J.Y.; Xing, S.J.; Chi, J.B.; Song, Y.M. Seed germination of Chionanthus retusa and Cedrela sinensis under salinity stress. J. Beijing For. Univ. 2003, 25, 80–83. [Google Scholar]
- Li, R.L.; Xu, B.M.; Sun, Y.T.; Guo, C.; Sun, C. Studies on Seed Domancy and Seed Gemination of Chionathus retusa and Rhodotypos Scandens in Beijing. Seed 2007, 29–31. [Google Scholar] [CrossRef]
- Chien, C.T.; Kuo-Huang, L.L.; Shen, Y.C.; Zhang, R.C.; Chen, S.Y.; Yang, J.C.; Pharis, R.P. Storage behavior of Chionanthus retusus seed and asynchronous development of the radicle and shoot apex during germination in relation to germination inhibitors, including abscisic acid and four phenolic glucosides. Plant Cell Physiol. 2004, 45, 1158–1167. [Google Scholar] [CrossRef]
- Niu, M.G.; Ren, J.; Li, J.H.; Dang, C.L.; Liu, C.S.; Liu, Y.; Sun, M.T.; Wang, J.Y. Comprehensive Evaluation of Seed and Fruit Traits in 44 Chionanthus retusus Germplasm Resources. Seed 2023, 42, 72–78. [Google Scholar]
- Tao, L. Distribution and Protection of Wild Plant Resources of Chionanthus retusus in Shanxi Province. Shanxi For. Sci. Technol. 2021, 50, 63–64. [Google Scholar]
- Qu, K.; Guo, H.P.; Wang, B.R.; Zhou, W.L.; Hou, L.L.; Li, Q.; Li, J.H.; Cheng, T.T. Genetic diversity analysis of Chionanthus retusus natural population based on SRAP molecular markers. J. Beijing For. Univ. 2020, 42, 40–50. [Google Scholar]
- Qu, K.; Li, J.H.; Guo, H.P.; Tian, Y.T.; Wang, B.R.; Hou, L.L.; Zhou, W.L. Investigation and Protection Strategies of Ancient Chionanthus retusus in Shandong Province. J. Shandong Agric. Univ. (Nat. Sci. Ed.) 2020, 51, 818–824. [Google Scholar]
- Yang, C.L.; Sun, F.Y.; Song, S.W.; Wang, F.M.; Zhuang, D.; Cui, Y.; Yin, Q.Y. Preliminary study on rescue technology of ancient and famous trees after disaster in Jining City—Taking the rescue and protection project of ancient Chionanthus retusus in Daizhuang Area as an example. For. By-Prod. Spec. China 2022, 31–32+40. [Google Scholar] [CrossRef]
- Wang, H.J. Preliminary study on the treatment and rejuvenation techniques of ancient fallen trees: A case study of ancient Chionanthus retusus in Jining City for more than 300 years. Land. Green. 2022, 55–57. [Google Scholar]
- Wang, J.N.; Xu, D.; Sang, Y.L.; Sun, M.T.; Liu, C.S.; Niu, M.G.; Li, Y.; Liu, L.S.; Han, X.J.; Li, J.H. A telomere-to-telomere gap-free reference genome of Chionanthus retusus provides insights into the molecular mechanism underlying petal shape changes. Hort. Res. 2024, uhae249. [Google Scholar] [CrossRef]
- Sun, M.T.; Wang, D.Y.; Li, Y.; Niu, M.G.; Liu, C.S.; Liu, L.S.; Wang, J.N.; Li, J.H. Genome-wide identification and expression pattern analysis of MIKC-Type MADS-box genes in Chionanthus retusus, an androdioecy plant. BMC Genom. 2024, 25, 662. [Google Scholar] [CrossRef]
- Wadl, P.A.; Rinehart, T.A.; Olsen, R.T.; Waldo, B.D.; Kirkbride, J.H. Genetic diversity and population structure of Chionanthus virginicus. J. Am. Soc. Hortic. Sci. 2022, 147, 62–69. [Google Scholar] [CrossRef]
- He, Y.X.; Liu, L.X.; Yang, S.H.; Dong, M.F.; Yuan, W.J.; Shang, F.D. Characterization of the complete chloroplast genome of Chinese fringetree (Chionanthus retusus). Conserv. Genet. Resour 2017, 9, 431–434. [Google Scholar] [CrossRef]
- Pan, B.P.; Bu, W.J. Progress in Genetic and Evolutionary Research on Mitochondrial Genomes. Biological Bulletin. 2005, 40, 1–3. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A fexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, research0082.1. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management andevolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Stefan, K.; Choudhuri, J.V.; Enno, O.; Chris, S.; Jens, S.; Robert, G. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C: CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly andcomparative analysis of the complete mitogenomes of Suaeda Glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, R. The complete mitochondrial genome of Osmanthus fragrans (Lamiales, Oleaceae) from China. Mitochondrial DNA Part B 2021, 6, 2056–2057. [Google Scholar] [CrossRef]
- Sadder, M.; Brake, M.; Ayoub, S.; Abusini, Y.; Al-Amad, I.; Haddad, N. Complete mitochondrial genome sequence of historical olive (Olea europaea Linnaeus 1753 subsp. europaea) cultivar Mehras in Jordan. Mitochondrial DNA Part B 2023, 8, 1205–1208. [Google Scholar] [CrossRef]
- Song, Y.; Du, X.; Li, A.; Fan, A.; He, L.; Sun, Z.; Niu, Y.; Qiao, Y. Assembly and analysis of the complete mitochondrial genome of Forsythia suspensa (Thunb.) Vahl. BMC Genom. 2023, 24, 708. [Google Scholar] [CrossRef]
- Qi, X.Y.; Wang, K.Q.; Yang, L.P.; Deng, Z.S.; Sun, Z.H. The complete mitogenome sequence of the corallily (Lilium pumilum) and the Lanzhoulily (Lilium davidii) in China. Open Life Sci. 2020, 15, 1060–1067. [Google Scholar] [CrossRef]
- Deng, L.K.; Li, Y.; Yu, J.N. RNA Editing Sites in Chloroplast Protein-coding Genes in Leaf White Mutant of Triticum aestivum. Chin. Bull. Bot. 2012, 47, 581–593. [Google Scholar]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Guo, W.; Zhu, A.; Fan, W.; Mower, J.P. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2017, 213, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, W.; Yu, X.; Zhang, X.; Zhang, Z.; Liu, Y.; Wang, W.; Tian, X. Insights into molecular structure, genome evolution and phylogenetic implication through mitochondrial genome sequence of Gleditsia sinensis. Sci. Rep. 2021, 11, 14850. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Yun, Q.; Xu, Y.; Zhao, G.; Liu, J.; Shi, S.; Chen, Z.; Jia, L. Comprehensive analysis of complete mitochondrial genome of Sapindus mukorossi Gaertn.: An important industrial oil tree species in China. Ind. Crop Prod. 2021, 174, 114210. [Google Scholar] [CrossRef]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 Genes Genomes Genet. 2018, 9, 549–559. [Google Scholar] [CrossRef]
- Chen, L.; Dong, X.; Huang, H.; Xu, H.; Rono, P.C.; Cai, X.; Hu, G. Assembly and comparative analysis of the initial complete mitochondrial genome of Primulina hunanensis (Gesneriaceae): A cave-dwelling endangered plant. BMC Genom. 2024, 25, 322. [Google Scholar] [CrossRef]
- Fang, Y.; Wu, H.; Zhang, T.; Yang, M.; Yin, Y.; Pan, L.; Yu, X.; Zhang, X.; Hu, S.; Al-Mssallem, I.S.; et al. A complete sequence and transcriptomic analyses of date palm (Phoenix dactylifera L.) mitochondrial genome. PLoS ONE. 2012, 7, e37164. [Google Scholar] [CrossRef]
- Zhang, D.; Li, W.X.; Gao, F.L.; Wang, G.T. Application of PhyloSuite to Phylogenetic Analysis Using Concatenated Sequences. Bio-Protoc. 2021, 101, e1010661. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Name of Genes |
|---|---|
| ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| NADH dehydrogenase | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 |
| Cytochrome b | cob |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFC, ccmFN |
| Cytochrome c oxidase | cox1, cox2, cox3 |
| Maturases | matR |
| Protein transport subunit | mttB |
| Ribosomal protein large subunit | rpl2, rpl5, rpl10, rpl16 |
| Ribosomal protein small subunit | rps3, rps4, rps7, rps10, rps12, rps13, rps14 |
| Succinate dehydrogenase | sdh3, sdh4 |
| Ribosome RNA | rrn5, rrn18, rrn26 |
| Transfer RNA | trnC-GCA (×3) 1, trnD-GUC (×3), trnE-UUC (×3), trnF-GAA, trnfM-CAU, trnG-GCC, trnHGUG, trnI-CAU (×2), trnK-UUU, trnM-CAU (×2), trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnS-CGA, trnS-GCU, trnS-GGA, trnS-UGA, trnTGGU, trnW-CCA (×2), trnY-GUA (×3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, S.; Lin, F.; Shu, X.; Niu, H.; Jing, Q.; Gao, L.; Gao, X.; Liu, D. Mitochondrial Genome Assembly and Comparative Analysis of Chionanthus Retusus (Oleaceae). Genes 2024, 15, 1523. https://doi.org/10.3390/genes15121523
Zhai S, Lin F, Shu X, Niu H, Jing Q, Gao L, Gao X, Liu D. Mitochondrial Genome Assembly and Comparative Analysis of Chionanthus Retusus (Oleaceae). Genes. 2024; 15(12):1523. https://doi.org/10.3390/genes15121523
Chicago/Turabian StyleZhai, Shasha, Furong Lin, Xiuge Shu, Hongyun Niu, Qi Jing, Lei Gao, Xiangbin Gao, and Dan Liu. 2024. "Mitochondrial Genome Assembly and Comparative Analysis of Chionanthus Retusus (Oleaceae)" Genes 15, no. 12: 1523. https://doi.org/10.3390/genes15121523
APA StyleZhai, S., Lin, F., Shu, X., Niu, H., Jing, Q., Gao, L., Gao, X., & Liu, D. (2024). Mitochondrial Genome Assembly and Comparative Analysis of Chionanthus Retusus (Oleaceae). Genes, 15(12), 1523. https://doi.org/10.3390/genes15121523