

High Expression of THY1 in Intestinal Gastric Cancer as a Key Factor in Tumor Biology: A Poor Prognosis-Independent Marker Related to the Epithelial–Mesenchymal Transition Profile

Abstract
:1. Introduction
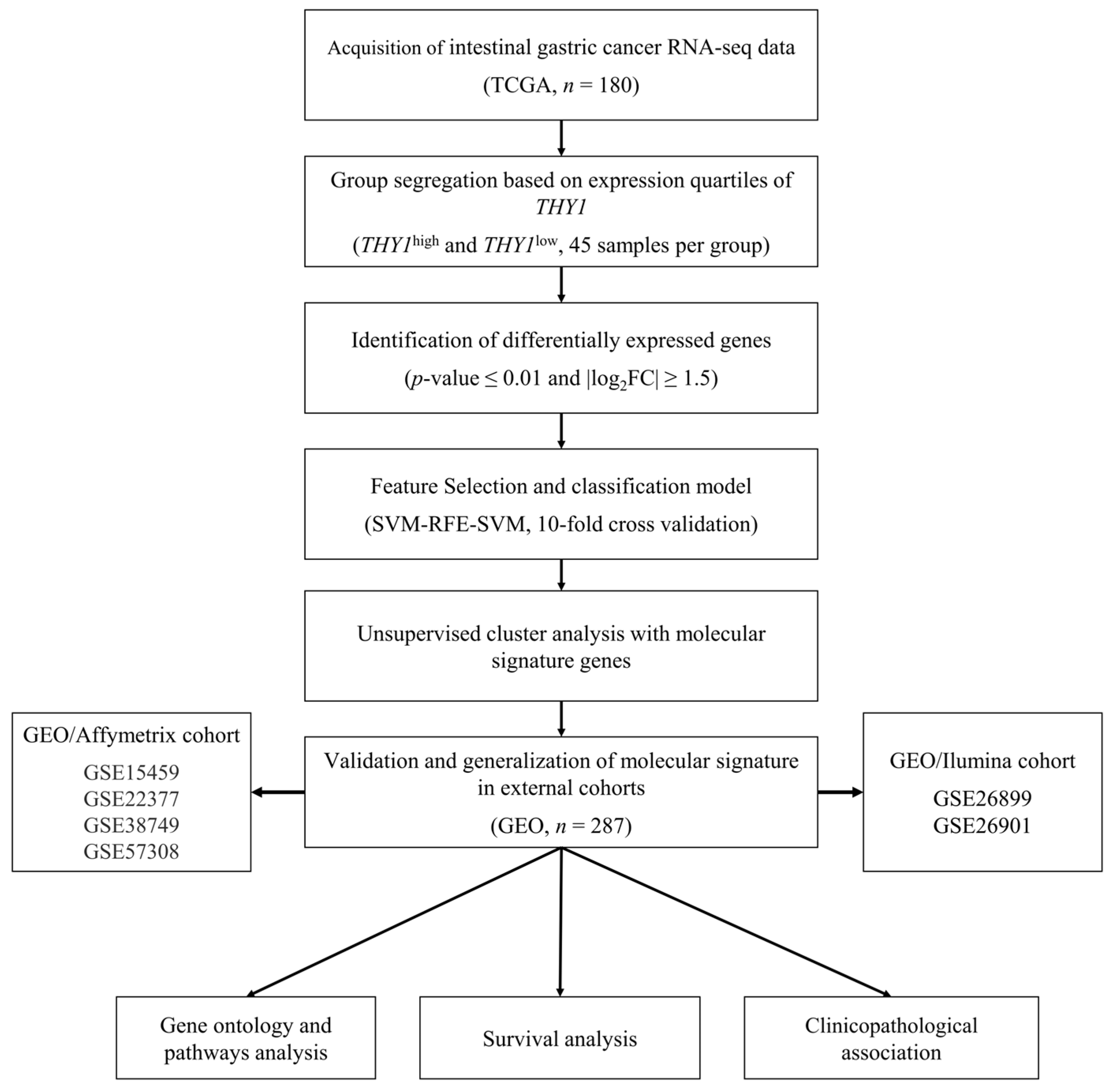
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. Segregation of Samples According to the Expression Level of THY1
2.3. Differential Gene Expression Analysis and Differentially Expressed Gene Identification
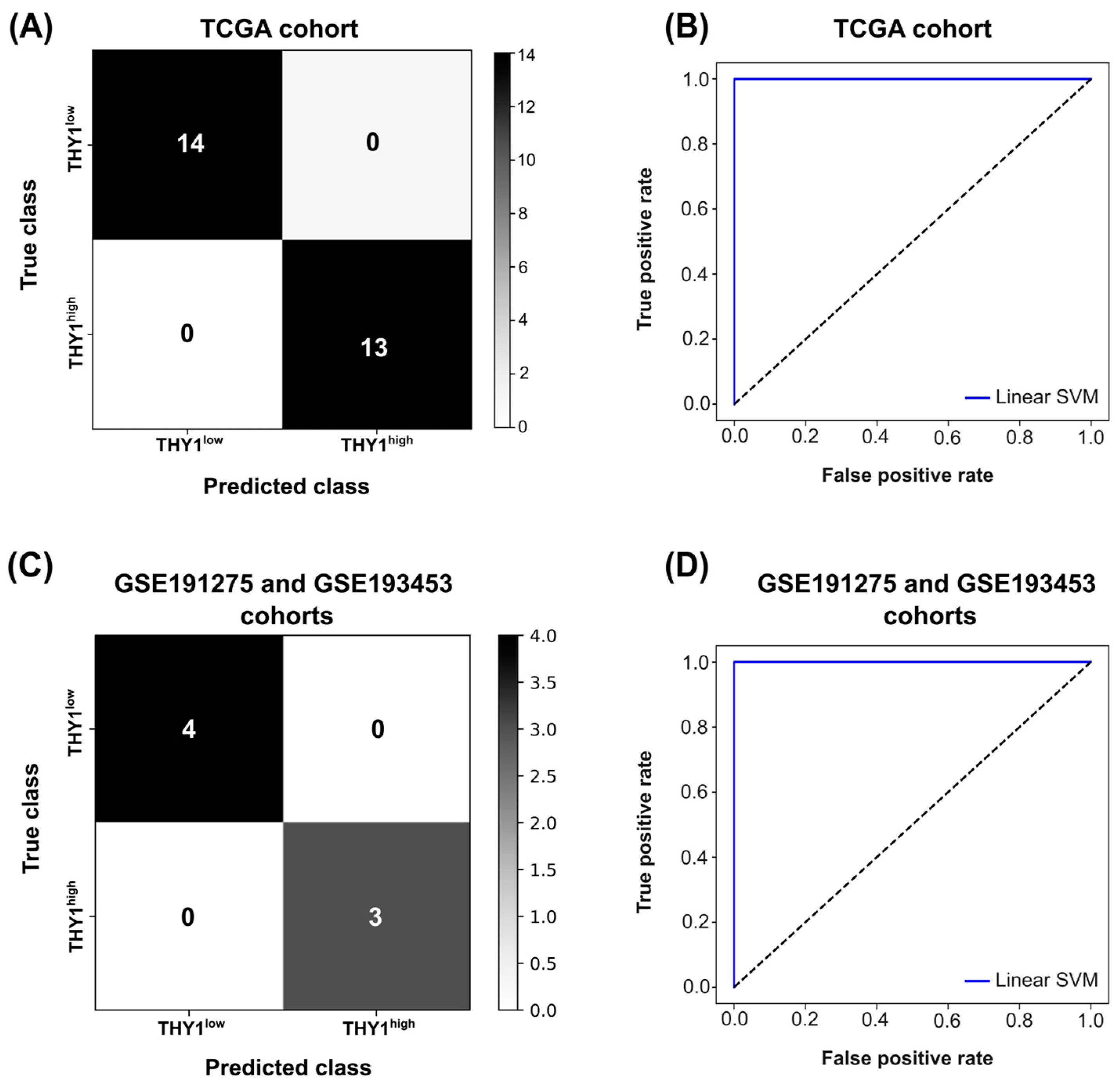
2.4. Feature Selection and Construction of a Supervised Machine Learning Model
2.5. Unsupervised Cluster Analysis
2.6. Molecular Signature Validation
2.7. Gene Ontology and Pathway Analysis
2.8. Survival Analysis and Clinicopathological Characteristics Association
2.9. Statistical Analysis
3. Results
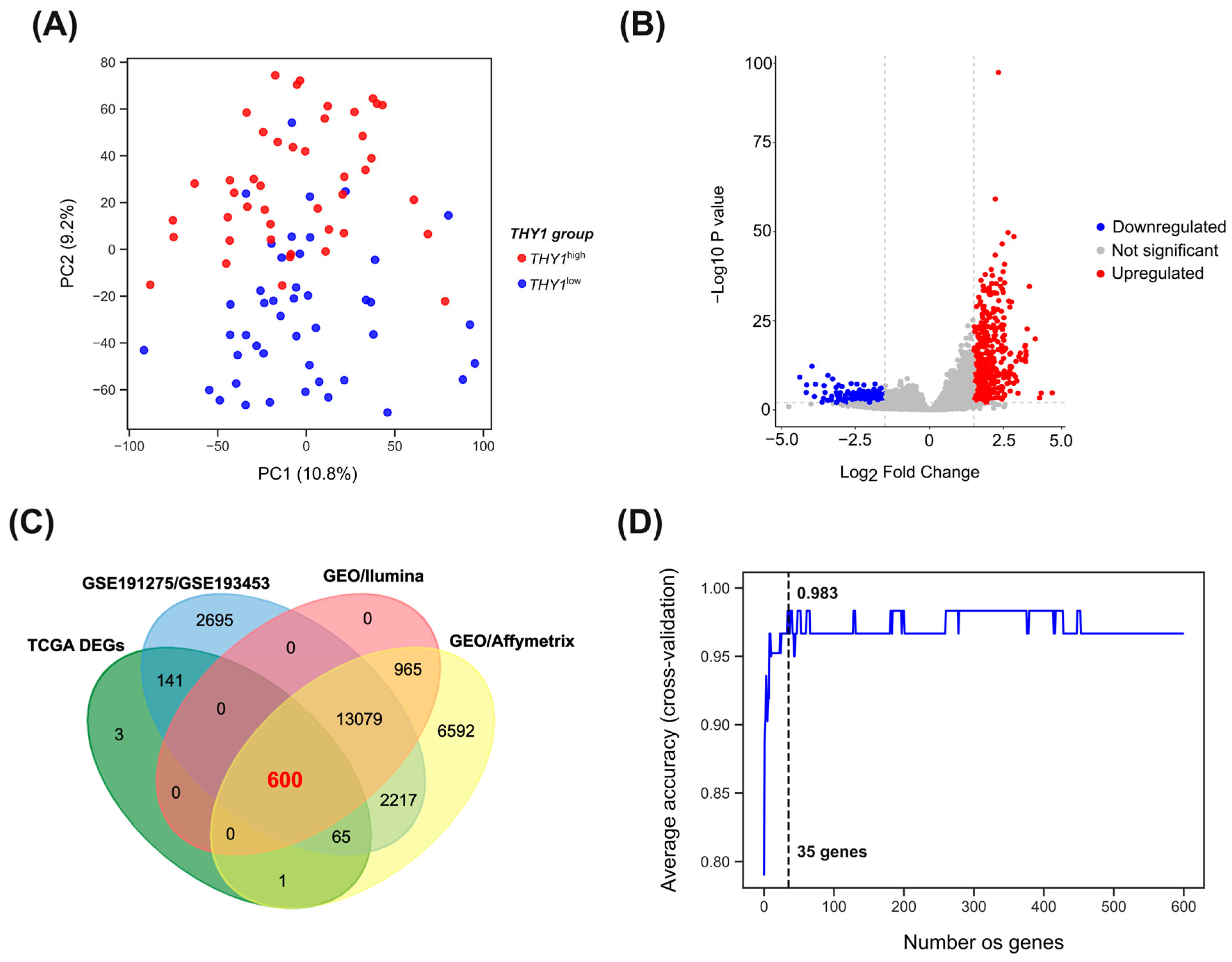
3.1. Segregation of THY1high and THY1low Groups
3.2. Differential Gene Expression between THY1high and THY1low Groups
3.3. Feature Selection and Supervised Machine Learning Approach Reveals a Group of Genes with High Discriminatory Power for THY1high IGC Tumors
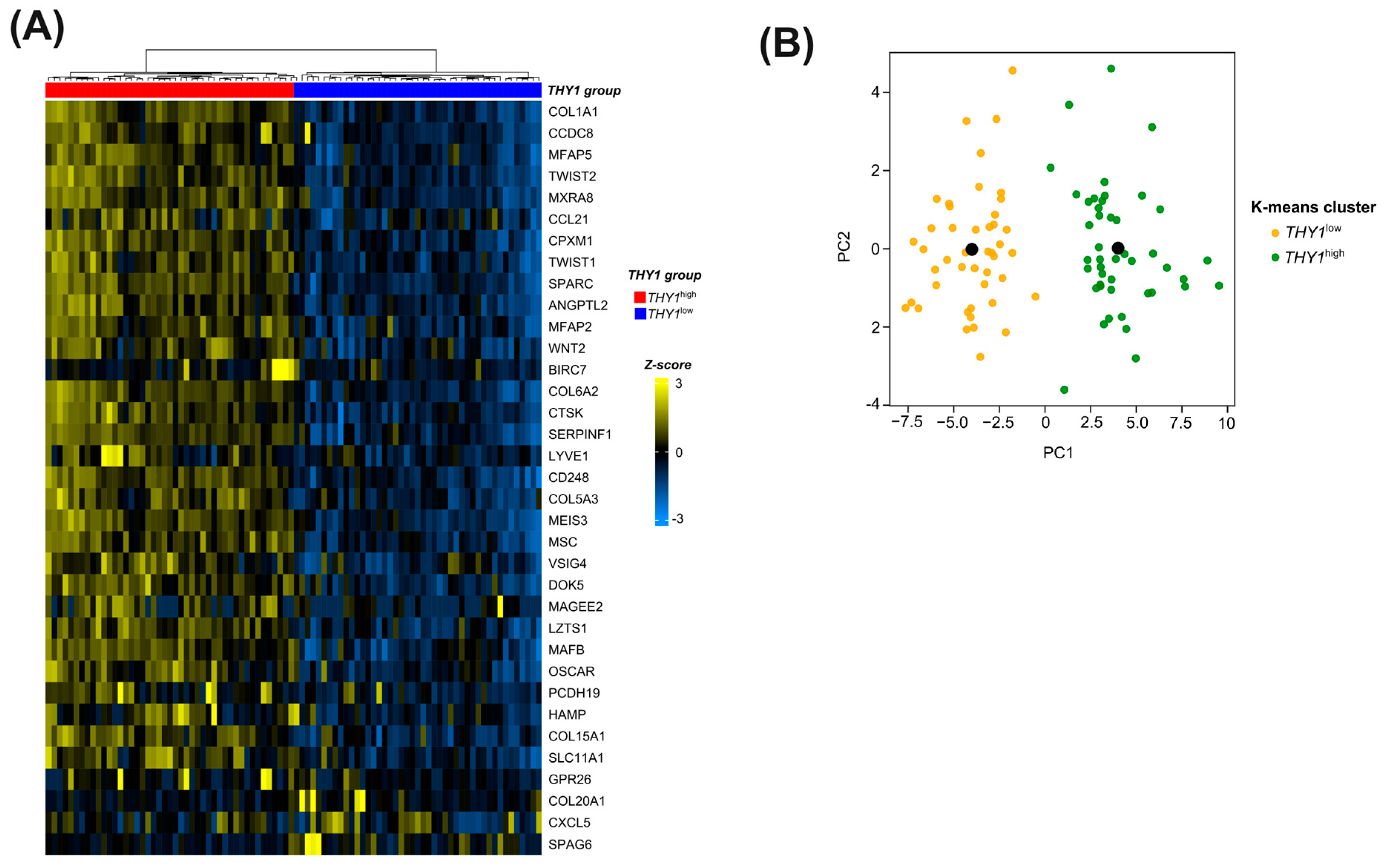
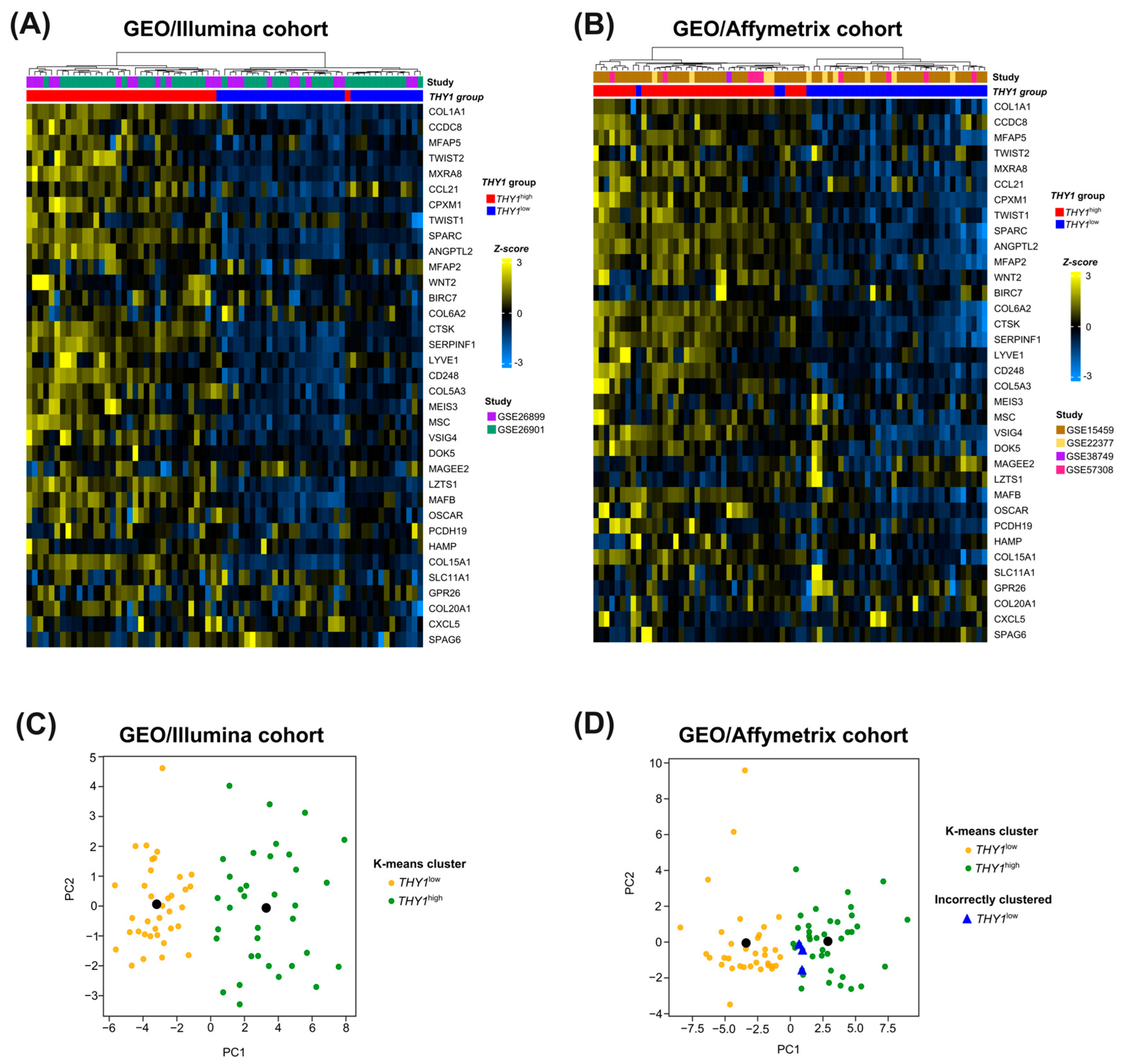
3.4. Candidate Genes in the Molecular Signature Can Successfully Segregate between THY1high and THY1low IGC Tumors in an Unsupervised Manner
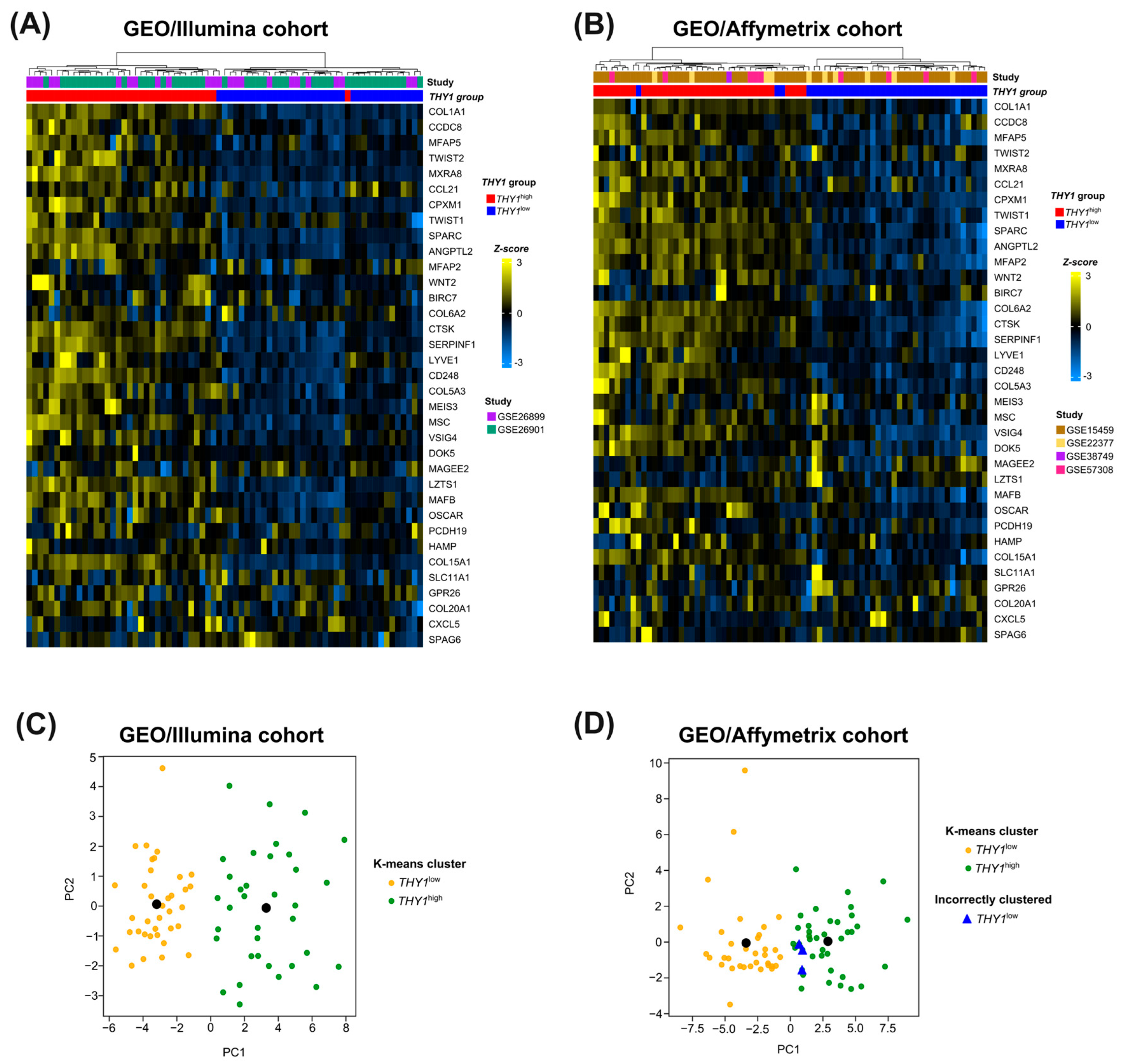
3.5. External Cohort Validation Reveals the Robustness and Generalization Capacity of the 35-Gene Molecular Signature for THY1high IGC Tumors
3.6. Molecular Signature Genes of THY1high IGC Tumors Are Involved in Key Processes of the Epithelial–Mesenchymal Transition and Remodeling of Tumor Tissue Composition
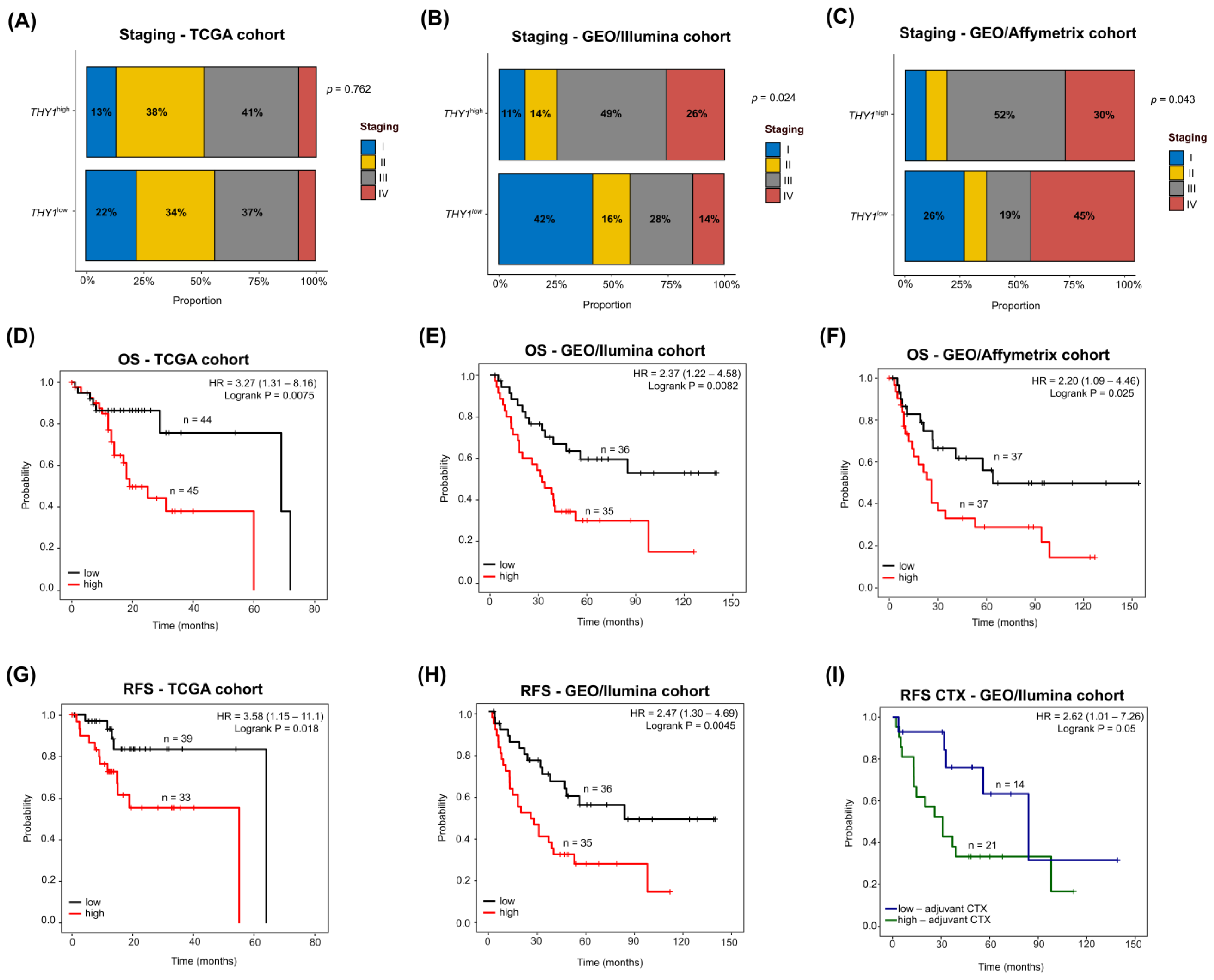
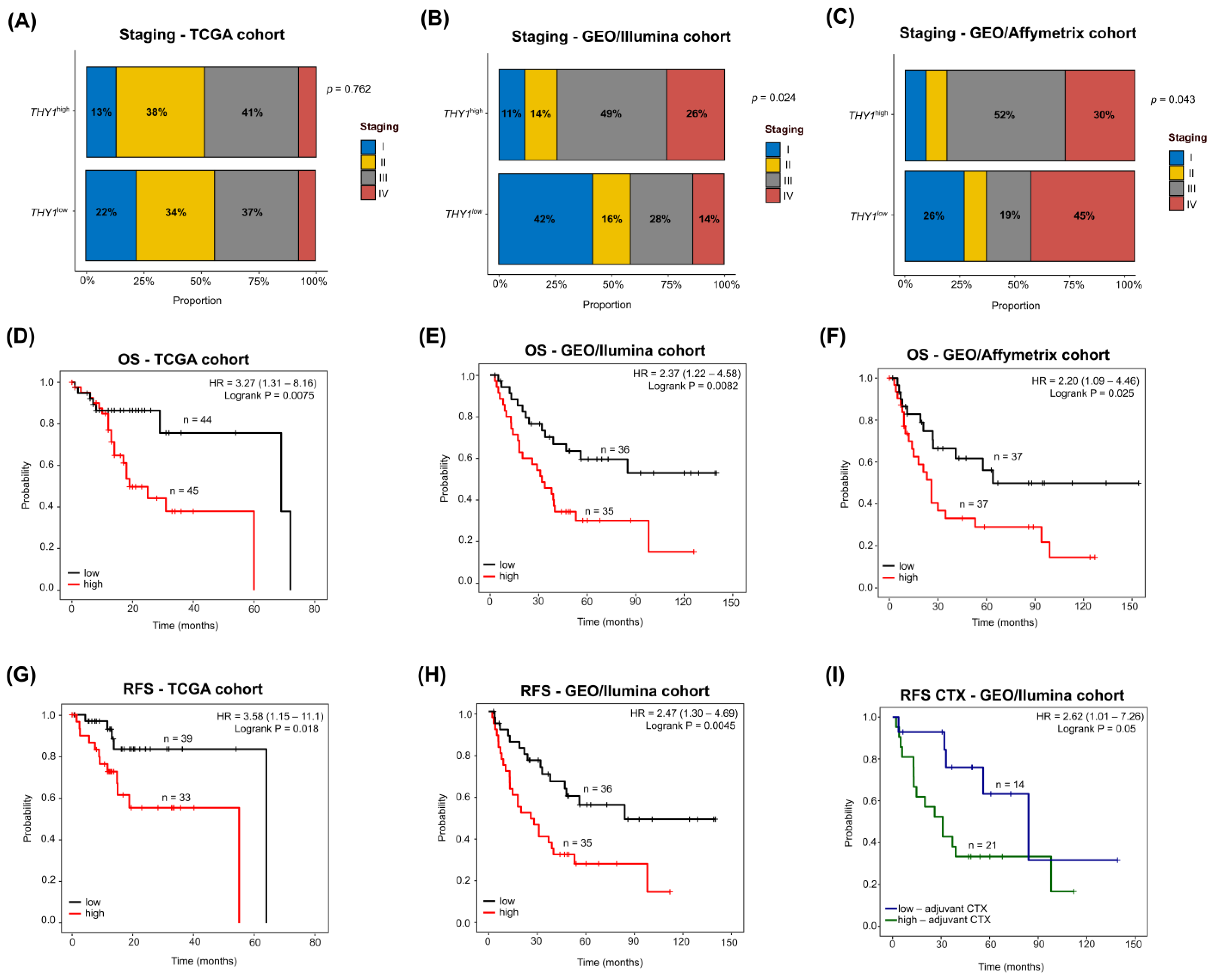
3.7. THY1high IGC Tumors Lead to Poor Survival with a Heterogeneous Clinicopathological Staging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Puia, A.; Schlanger, D.; Fetti, A.; Popa, C.; Necula, A.; Al Hajjar, N. Do Patient Delay and Diagnostic Delay Influence Long Term Survival in Patients with Gastric Cancer? J. Gastrointestin Liver Dis. 2022, 31, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Witzig, R.; Schönberger, B.; Fink, U.; Busch, R.; Gundel, H.; Sendler, A.; Peschel, C.; Siewert, J.R.; Lordick, F. Delays in diagnosis and therapy of gastric cancer and esophageal adenocarcinoma. Endoscopy 2006, 38, 1122–1126. [Google Scholar] [CrossRef]
- Feng, X.Y.; Chen, Y.B.; Wang, W.; Guan, Y.X.; Li, Y.F.; Chen, S.; Sun, X.W.; Li, W.; Xu, D.Z.; Zhan, Y.Q.; et al. Time-varying pattern of recurrence risk for gastric cancer patients. Med. Oncol. 2013, 30, 514. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.K.; Raj, S.S.; Shankar, E.M.; Ganapathy, E.; Ebrahim, A.S.; Farooq, S.M. Gastric Carcinoma: A Review on Epidemiology, Current Surgical and Chemotherapeutic Options [Internet]. Gastric Carcinoma-New Insights into Current Management. IntechOpen. 2013. Available online: https://www.intechopen.com/state.item.id (accessed on 21 December 2022).
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef]
- Tang, C.T.; Zeng, L.; Yang, J.; Zeng, C.; Chen, Y. Analysis of the Incidence and Survival of Gastric Cancer Based on the Lauren Classification: A Large Population-Based Study Using SEER. Front. Oncol. 2020, 10, 1212. [Google Scholar] [CrossRef]
- Parsonnet, J.; Vandersteen, D.; Goates, J.; Sibley, R.K.; Pritikin, J.; Chang, Y. Helicobacter pylori Infection in Intestinal- and Diffuse-Type Gastric Adenocarcinomas. JNCI J. Natl. Cancer Inst. 1991, 83, 640–643. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Correa, P. A human model of gastric carcinogenesis. Cancer Res. 1988, 48, 3554–3560. [Google Scholar] [PubMed]
- Binato, R.; Santos, E.C.; Boroni, M.; Demachki, S.; Assumpção, P.; Abdelhay, E. A common molecular signature of intestinal-type gastric carcinoma indicates processes related to gastric carcinogenesis. Oncotarget 2018, 9, 7359–7371. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, K.; Chen, X.; Chen, R.; Zou, Y. Bioinformatics analysis identifies COL1A1, THBS2 and SPP1 as potential predictors of patient prognosis and immunotherapy response in gastric cancer. Biosci. Rep. 2021, 41, BSR20202564. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chung, H.C. Novel biomarker candidates for gastric cancer. Oncol. Rep. 2008, 19, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qu, Y.; Zhou, H.; Zheng, Z.; Zhao, J.; Zhang, J. Bioinformatic analysis of potential hub genes in gastric adenocarcinoma. Sci. Prog. 2021, 104, 00368504211004260. [Google Scholar] [CrossRef] [PubMed]
- Kroczek, R.A.; Gunter, K.C.; Germain, R.N.; Shevach, E.M. Thy-1 functions as a signal transduction molecule in T lymphocytes and transfected B lymphocytes. Nature 1986, 322, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Morris, R. Thy-1 in developing nervous tissue. Dev. Neurosci. 1985, 7, 133–160. [Google Scholar] [CrossRef]
- Zhou, Y.; Hagood, J.S.; Lu, B.; Merryman, W.D.; Murphy-Ullrich, J.E. Thy-1-integrin alphav beta5 interactions inhibit lung fibroblast contraction-induced latent transforming growth factor-beta1 activation and myofibroblast differentiation. J. Biol. Chem. 2010, 285, 22382–22393. [Google Scholar] [CrossRef]
- Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic catch of a Thy-1-α5β1+syndecan-4 trimolecular complex. Nat. Commun. 2014, 5, 4886. [Google Scholar] [CrossRef]
- Leyton, L.; Schneider, P.; Labra, C.V.; Rüegg, C.; Hetz, C.A.; Quest, A.F.; Bron, C. Thy-1 binds to integrin β3 on astrocytes and triggers formation of focal contact sites. Curr. Biol. 2001, 11, 1028–1038. [Google Scholar] [CrossRef]
- Choi, J.; Leyton, L.; Nham, S.U. Characterization of alphaX I-domain binding to Thy-1. Biochem. Biophys. Res. Commun. 2005, 331, 557–561. [Google Scholar] [CrossRef]
- Wetzel, A.; Chavakis, T.; Preissner, K.T.; Sticherling, M.; Haustein, U.F.; Anderegg, U.; Saalbach, A. Human Thy-1 (CD90) on activated endothelial cells is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Immunol. 2004, 172, 3850–3859. [Google Scholar] [CrossRef] [PubMed]
- Beech, J.N.; Morris, R.J.; Raisman, G. Density of Thy-1 on Axonal Membrane of Different Rat Nerves. J. Neurochem. 1983, 41, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.; Kay, R.; Cutler, R.L.; Lansdorp, P.M. Expression of Thy-1 on human hematopoietic progenitor cells. J. Exp. Med. 1993, 177, 1331–1342. [Google Scholar] [CrossRef]
- Killeen, N. T-cell regulation: Thy-1–hiding in full view. Curr. Biol. 1997, 7, R774–R777. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.M.; Currie, I.S.; Terrace, J.D.; Garden, O.J.; Parks, R.W.; Ross, J.A. Hepatic progenitor cells in human fetal liver express the oval cell marker Thy-1. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 291, G45–G54. [Google Scholar] [CrossRef]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705. [Google Scholar] [CrossRef]
- Abeysinghe, H.R.; Cao, Q.; Xu, J.; Pollock, S.; Veyberman, Y.; Guckert, N.L.; Keng, P.; Wang, N. THY1 expression is associated with tumor suppression of human ovarian cancer. Cancer Genet. Cytogenet. 2003, 143, 125–132. [Google Scholar] [CrossRef]
- Chen, L.; Chau, W.Y.; Yuen, H.T.; Liu, X.H.; Qi, R.Z.; Lung, M.L.; Lung, H.L. THY1 (CD90) Maintains the Adherens Junctions in Nasopharyngeal Carcinoma via Inhibition of SRC Activation. Cancers 2023, 15, 2189. [Google Scholar] [CrossRef]
- Hu, Y.; Jin, D.; Zhou, Y.; Cheng, Y.; Cao, H.; Ma, Y.; Zhang, W. Multiple roles of THY1 in gastric cancer based on data mining. Transl. Cancer Res. 2020, 9, 2748–2757. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Liu, H.; Pan, Y.; Sun, L.; Yu, L.; Sun, L.; Yang, Z.; Ran, Y. Distinct biological characterization of the CD44 and CD90 phenotypes of cancer stem cells in gastric cancer cell lines. Mol. Cell Biochem. 2019, 459, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Zhang, Z.Z.; Xu, J.; Wang, M.; Zhu, C.C.; Zhuang, C.; Liu, Q.; Zhao, G.; Cao, H. THY-1 (CD90) expression promotes the growth of gastric cancer cells. Int. J. Clin. Exp. Pathol. 2017, 10, 9878–9888. [Google Scholar] [PubMed]
- Zhu, G.C.; Gao, L.; He, J.; Long, Y.; Liao, S.; Wang, H.; Li, X.; Yi, W.; Pei, Z.; Wu, M.; et al. CD90 is upregulated in gastric cancer tissues and inhibits gastric cancer cell apoptosis by modulating the expression level of SPARC protein. Oncol. Rep. 2015, 34, 2497–2506. [Google Scholar] [CrossRef]
- Gao, L.; Li, J.; He, J.; Liang, L.; He, Z.; Yue, C.; Jin, X.; Luo, G.; Zhou, Y. CD90 affects the biological behavior and energy metabolism level of gastric cancer cells by targeting the PI3K/AKT/HIF-1α signaling pathway. Oncol. Lett. 2021, 21, 191. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- LaValle, S.M.; Branicky, M.S.; Lindemann, S.R. On the Relationship between Classical Grid Search and Probabilistic Roadmaps. Int. J. Robot. Res. 2004, 23, 673–692. [Google Scholar] [CrossRef]
- Ting, K.M. Confusion Matrix. In Encyclopedia of Machine Learning and Data Mining; Sammut, C., Webb, G.I., Eds.; Springer: Boston, MA, USA, 2017; p. 260. [Google Scholar] [CrossRef]
- Hajian-Tilaki, K. Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Casp. J. Intern. Med. 2013, 4, 627–635. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009; 242p. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation [Internet] 2021; Volume 2. Available online: https://www.cell.com/the-innovation/abstract/S2666-6758(21)00066-7 (accessed on 23 November 2023).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Oh, S.C.; Sohn, B.H.; Cheong, J.H.; Kim, S.B.; Lee, J.E.; Park, K.C.; Lee, S.H.; Park, J.L.; Park, Y.Y.; Lee, H.S.; et al. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat. Commun. 2018, 9, 1777. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Chi, C.; Liu, T.; Yang, S.; Wang, B.; Han, W.; Li, J. ISLR affects colon cancer progression by regulating the epithelial-mesenchymal transition signaling pathway. Anticancer. Drugs 2022, 33, e670–e679. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Pocard, M.; Mirshahi, M. Targeting the differentiation of gastric cancer cells (KATO-III) downregulates epithelial-mesenchymal and cancer stem cell markers. Oncol. Rep. 2019, 42, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo-Lozano, N.; Alcalá, S.; Sainz, B.; Perona, R.; Sanchez-Perez, I. Targeting MAD2 modulates stemness and tumorigenesis in human Gastric Cancer cell lines. Theranostics 2020, 10, 9601–9618. [Google Scholar] [CrossRef]
- Aguilera, K.Y.; Rivera, L.B.; Hur, H.; Carbon, J.G.; Toombs, J.E.; Goldstein, C.D.; Dellinger, M.T.; Castrillon, D.H.; Brekken, R.A. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014, 74, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef]
- Díaz Del Arco, C.; Estrada Muñoz, L.; Ortega Medina, L.; Molina Roldán, E.; Cerón Nieto, M.Á.; García Gómez de Las Heras, S.; Aceñero, M.J.F. Clinicopathological differences, risk factors and prognostic scores for western patients with intestinal and diffuse-type gastric cancer. World J. Gastrointest. Oncol. 2022, 14, 1162–1174. [Google Scholar] [CrossRef]
- Hu, P.; Barker, T.H. Thy-1 in Integrin Mediated Mechanotransduction. Front. Cell Dev. Biol. 2019, 7, 22. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Debaugnies, M.; Rodríguez-Acebes, S.; Blondeau, J.; Parent, M.A.; Zocco, M.; Song, Y.; de Maertelaer, V.; Moers, V.; Latil, M.; Dubois, C.; et al. RHOJ controls EMT-associated resistance to chemotherapy. Nature 2023, 616, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Marfels, C.; Hoehn, M.; Wagner, E.; Günther, M. Characterization of in vivo chemoresistant human hepatocellular carcinoma cells with transendothelial differentiation capacities. BMC Cancer 2013, 13, 176. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Description | L2FC | p-Value | Expression in THY1high |
|---|---|---|---|---|
| COL1A1 | collagen type I α 1 chain | 2.85 | 2.53 × 10−49 | Upregulated |
| CCDC8 | coiled-coil domain-containing 8 | 2.68 | 3.03 × 10−31 | Upregulated |
| MFAP5 | Microfibril-associated protein 5 | 2.51 | 2.78 × 10−20 | Upregulated |
| TWIST2 | twist family bHLH transcription factor 2 | 2.34 | 2.32 × 10−31 | Upregulated |
| MXRA8 | matrix remodeling-associated 8 | 2.31 | 6.94 × 10−29 | Upregulated |
| CCL21 | C-C motif chemokine ligand 21 | 2.28 | 2.48 × 10−10 | Upregulated |
| CPXM1 | carboxypeptidase X, M14 family member 1 | 2.25 | 3.74 × 10−36 | Upregulated |
| TWIST1 | twist family bHLH transcription factor 1 | 2.22 | 1.03 × 10−21 | Upregulated |
| SPARC | secreted protein acidic and cysteine-rich | 2.22 | 6.93 × 10−60 | Upregulated |
| ANGPTL2 | Angiopoietin-like 2 | 2.16 | 3.30 × 10−29 | Upregulated |
| MFAP2 | microfibril-associated protein 2 | 2.13 | 4.23 × 10−17 | Upregulated |
| WNT2 | Wnt family member 2 | 2.11 | 1.74 × 10−13 | Upregulated |
| BIRC7 | baculoviral IAP repeat-containing 7 | 2.10 | 6.36 × 10−6 | Upregulated |
| COL6A2 | collagen type VI α 2 chain | 2.09 | 3.87 × 10−40 | Upregulated |
| CTSK | cathepsin K | 2.05 | 5.71 × 10−34 | Upregulated |
| SERPINF1 | serpin family F member 1 | 2.02 | 2.23 × 10−27 | Upregulated |
| LYVE1 | lymphatic vessel endothelial hyaluronan receptor 1 | 1.99 | 9.68 × 10−12 | Upregulated |
| CD248 | CD248 molecule | 1.88 | 1.06 × 10−38 | Upregulated |
| COL5A3 | collagen type V α 3 chain | 1.84 | 7.34 × 10−21 | Upregulated |
| MEIS3 | Meis homeobox 3 | 1.82 | 3.00 × 10−34 | Upregulated |
| MSC | musculin | 1.81 | 3.30 × 10−29 | Upregulated |
| VSIG4 | V-set and immunoglobulin domain-containing 4 | 1.80 | 5.93 × 10−10 | Upregulated |
| DOK5 | docking protein 5 | 1.75 | 1.56 × 10−18 | Upregulated |
| MAGEE2 | MAGE family member E2 | 1.72 | 9.09 × 10−4 | Upregulated |
| LZTS1 | leucine zipper tumor suppressor 1 | 1.70 | 9.92 × 10−21 | Upregulated |
| MAFB | MAF bZIP transcription factor B | 1.63 | 1.02 × 10−17 | Upregulated |
| OSCAR | osteoclast-associated Ig-like receptor | 1.60 | 2.33 × 10−13 | Upregulated |
| PCDH19 | protocadherin 19 | 1.60 | 1.71 × 10−6 | Upregulated |
| HAMP | hepcidin antimicrobial peptide | 1.54 | 2.17 × 10−5 | Upregulated |
| COL15A1 | collagen type XV α 1 chain | 1.53 | 2.96 × 10−17 | Upregulated |
| SLC11A1 | solute carrier family 11 member 1 | 1.52 | 4.84 × 10−14 | Upregulated |
| GPR26 | G protein-coupled receptor 26 | 1.51 | 6.34 × 10−3 | Upregulated |
| COL20A1 | collagen type XX α 1 chain | −1.69 | 6.09 × 10−5 | Downregulated |
| CXCL5 | C-X-C motif chemokine ligand 5 | −1.98 | 2.67 × 10−3 | Downregulated |
| SPAG6 | sperm-associated antigen 6 | −2.44 | 5.66 × 10−8 | Downregulated |
| Overrepresentation Analysis | Signature Genes | p-Value | Database | |
|---|---|---|---|---|
| Up | Down | |||
| Extracellular matrix organization | COL1A1, MFAP5, SPARC, MFAP2, COL6A2, CTSK, COL5A3, COL15A1 | COL20A1 | 7.19 × 10−9 | Reactome (R-HSA-1474244) |
| Collagen formation | COL1A1, COL6A2, COL5A3, COL15A1 | COL20A1 | 1.17 × 10−6 | Reactome (R-HSA-1474290) |
| Epithelial–mesenchymal transition | COL1A1, MFAP5, SPARC, COL5A3 | 2.77 × 10−6 | Hallmarks | |
| Positive regulation of cell migration | COL1A1, TWIST2, CCL21, TWIST1, SPARC, LYVE1 | 2.98 × 10−4 | GOBP (GO:0030335) | |
| Positive regulation of cell motility | COL1A1, TWIST2, CCL21, TWIST1, SPARC, LYVE1 | 3.74 × 10−4 | GOBP (GO:2000147) | |
| Positive regulation of locomotion | COL1A1, TWIST2, CCL21, TWIST1, SPARC, LYVE1 | 4.10 × 10−4 | GOBP (GO:0040017) | |
| Embryo development | COL1A1, MFAP5, TWIST1, MFAP2, MEIS3 | 2.18 × 10−3 | GOBP (GO:0009790) | |
| Epithelial–mesenchymal transition in colorectal cancer | TWIST2, TWIST1, SPARC | 2.44 × 10−3 | WikiPathways (WP4239) | |
| Positive regulation of epithelial–mesenchymal transition | COL1A1, TWIST1 | 5.04 × 10−3 | GOBP (GO:0010718) | |
| Regulation of angiogenesis | TWIST1, SPARC, SERPINF1 | 1.22 × 10−2 | GOBP (GO:0045765) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohan, P.; dos Santos, E.C.; Abdelhay, E.; Binato, R. High Expression of THY1 in Intestinal Gastric Cancer as a Key Factor in Tumor Biology: A Poor Prognosis-Independent Marker Related to the Epithelial–Mesenchymal Transition Profile. Genes 2024, 15, 28. https://doi.org/10.3390/genes15010028
Rohan P, dos Santos EC, Abdelhay E, Binato R. High Expression of THY1 in Intestinal Gastric Cancer as a Key Factor in Tumor Biology: A Poor Prognosis-Independent Marker Related to the Epithelial–Mesenchymal Transition Profile. Genes. 2024; 15(1):28. https://doi.org/10.3390/genes15010028
Chicago/Turabian StyleRohan, Paulo, Everton Cruz dos Santos, Eliana Abdelhay, and Renata Binato. 2024. "High Expression of THY1 in Intestinal Gastric Cancer as a Key Factor in Tumor Biology: A Poor Prognosis-Independent Marker Related to the Epithelial–Mesenchymal Transition Profile" Genes 15, no. 1: 28. https://doi.org/10.3390/genes15010028
APA StyleRohan, P., dos Santos, E. C., Abdelhay, E., & Binato, R. (2024). High Expression of THY1 in Intestinal Gastric Cancer as a Key Factor in Tumor Biology: A Poor Prognosis-Independent Marker Related to the Epithelial–Mesenchymal Transition Profile. Genes, 15(1), 28. https://doi.org/10.3390/genes15010028