

Epigenetic Findings in Twins with Esophageal Atresia

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Methods
2.3. Data Processing
2.4. Statistical Analyses
3. Results
3.1. Quality Check and Filtering
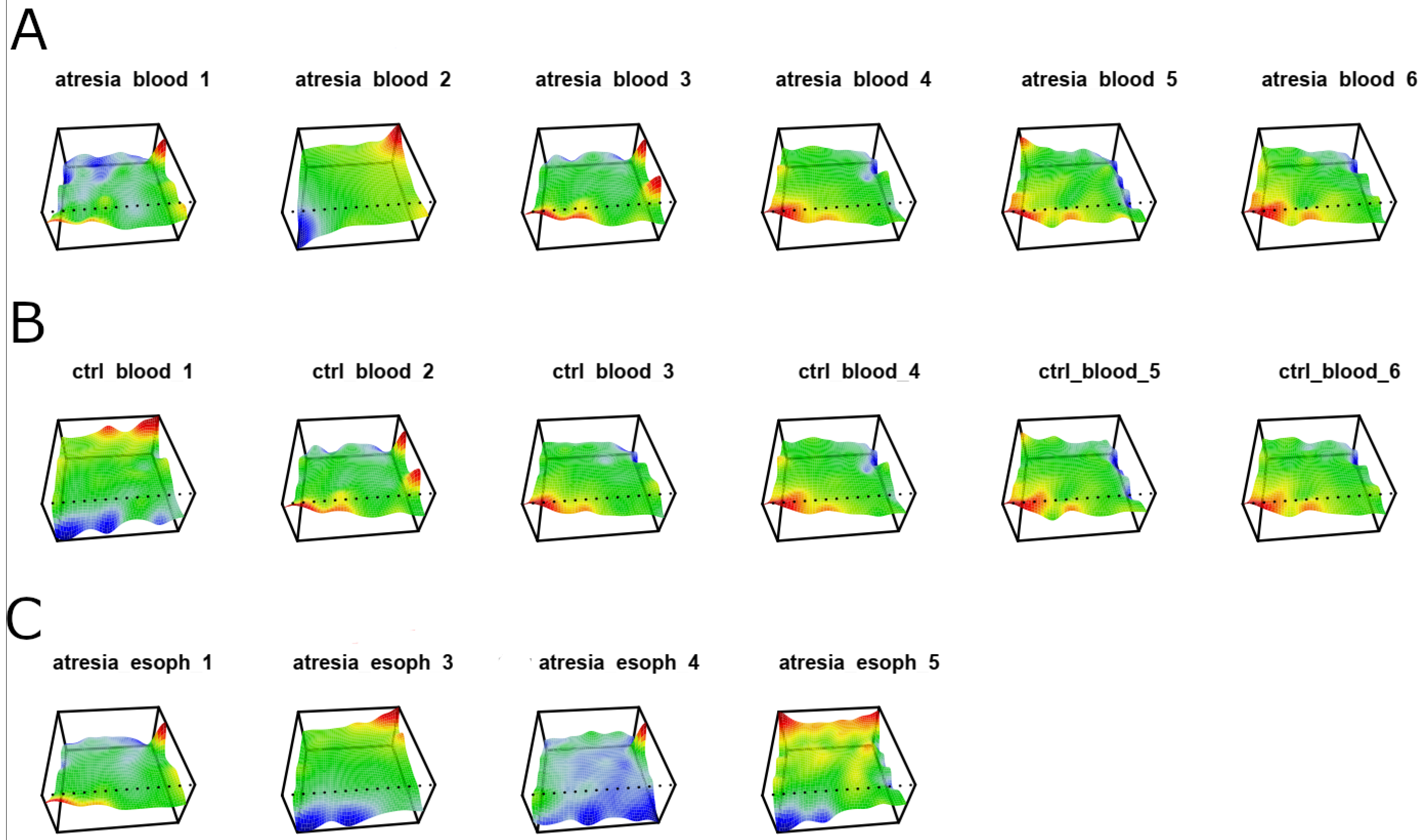
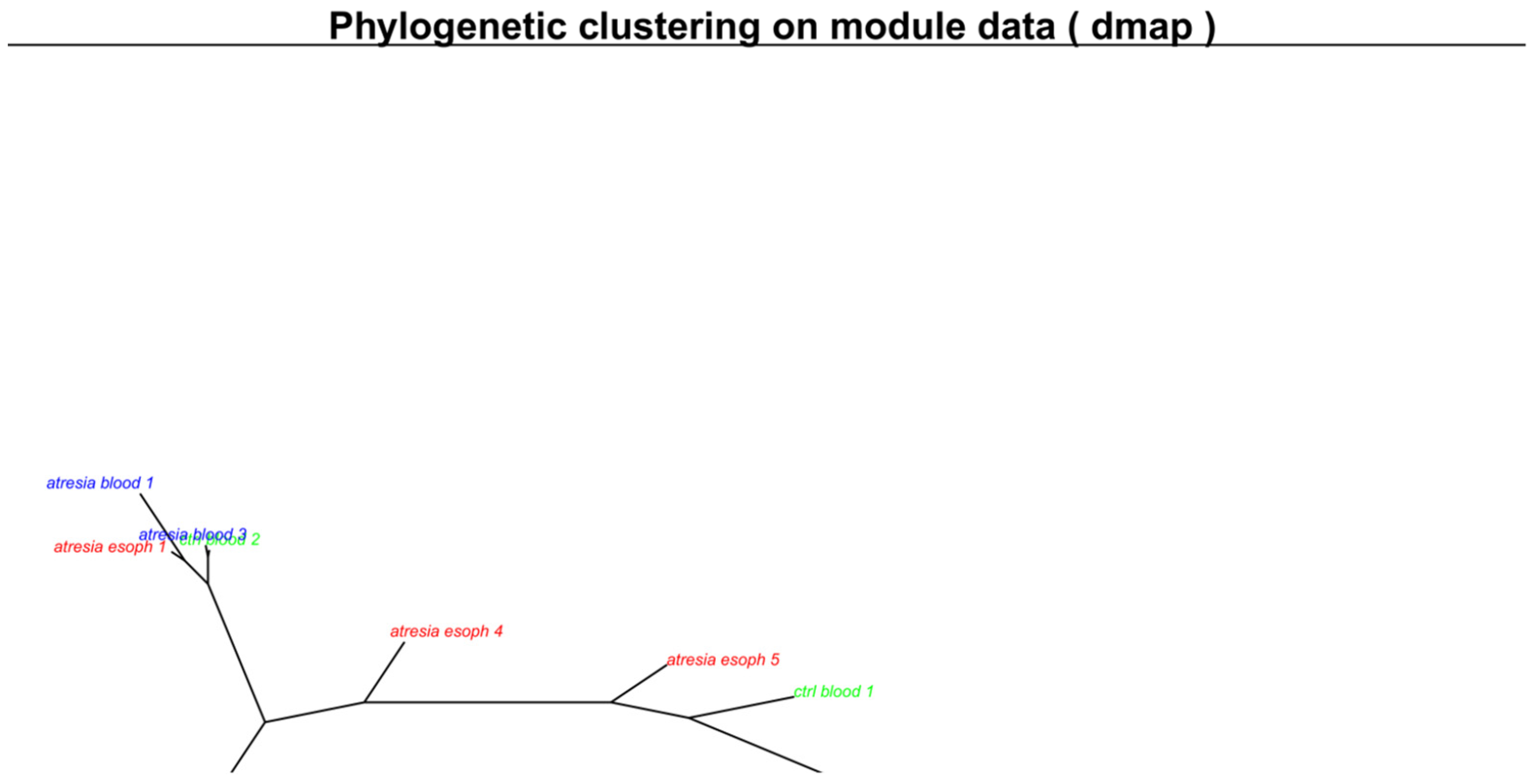
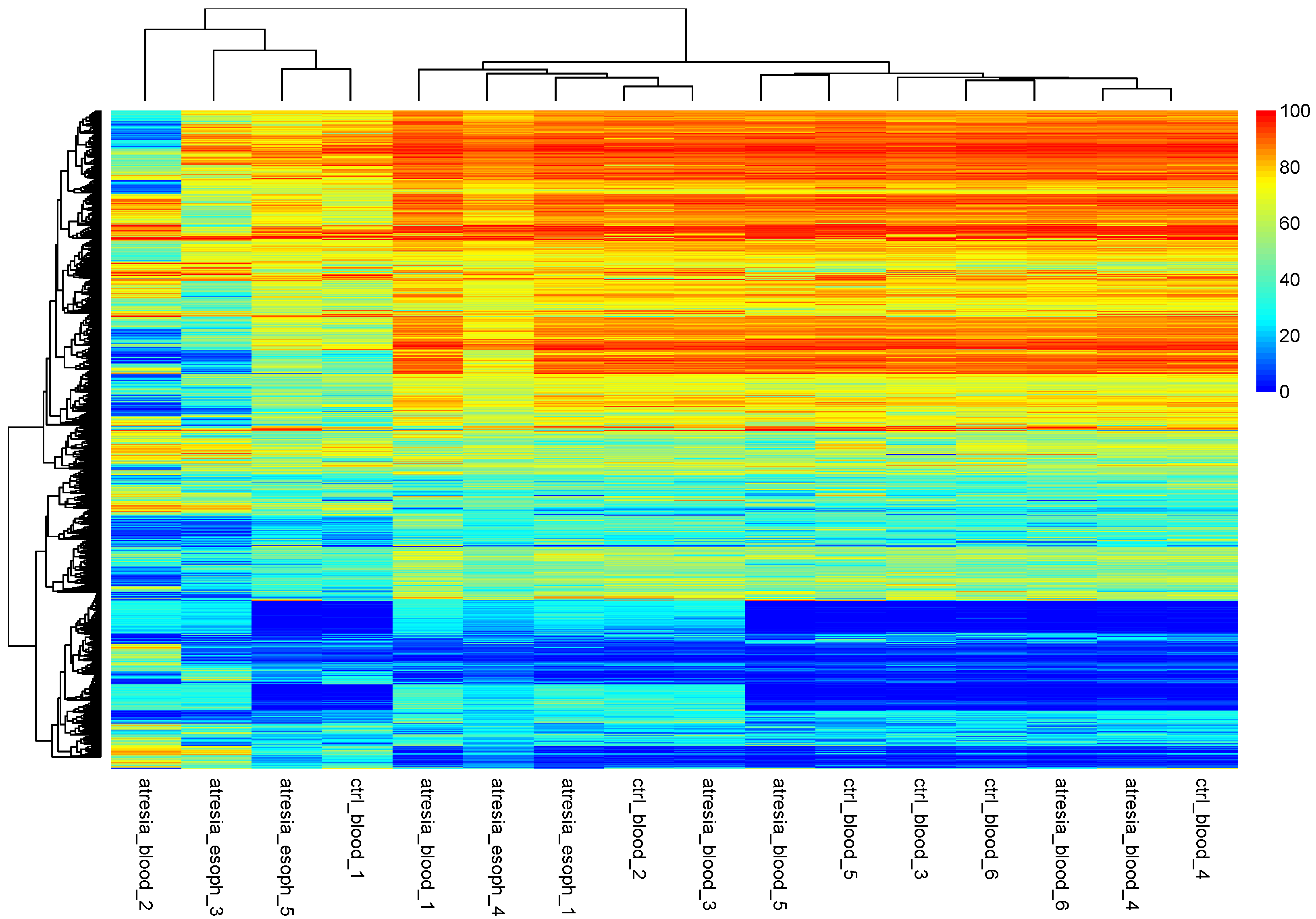
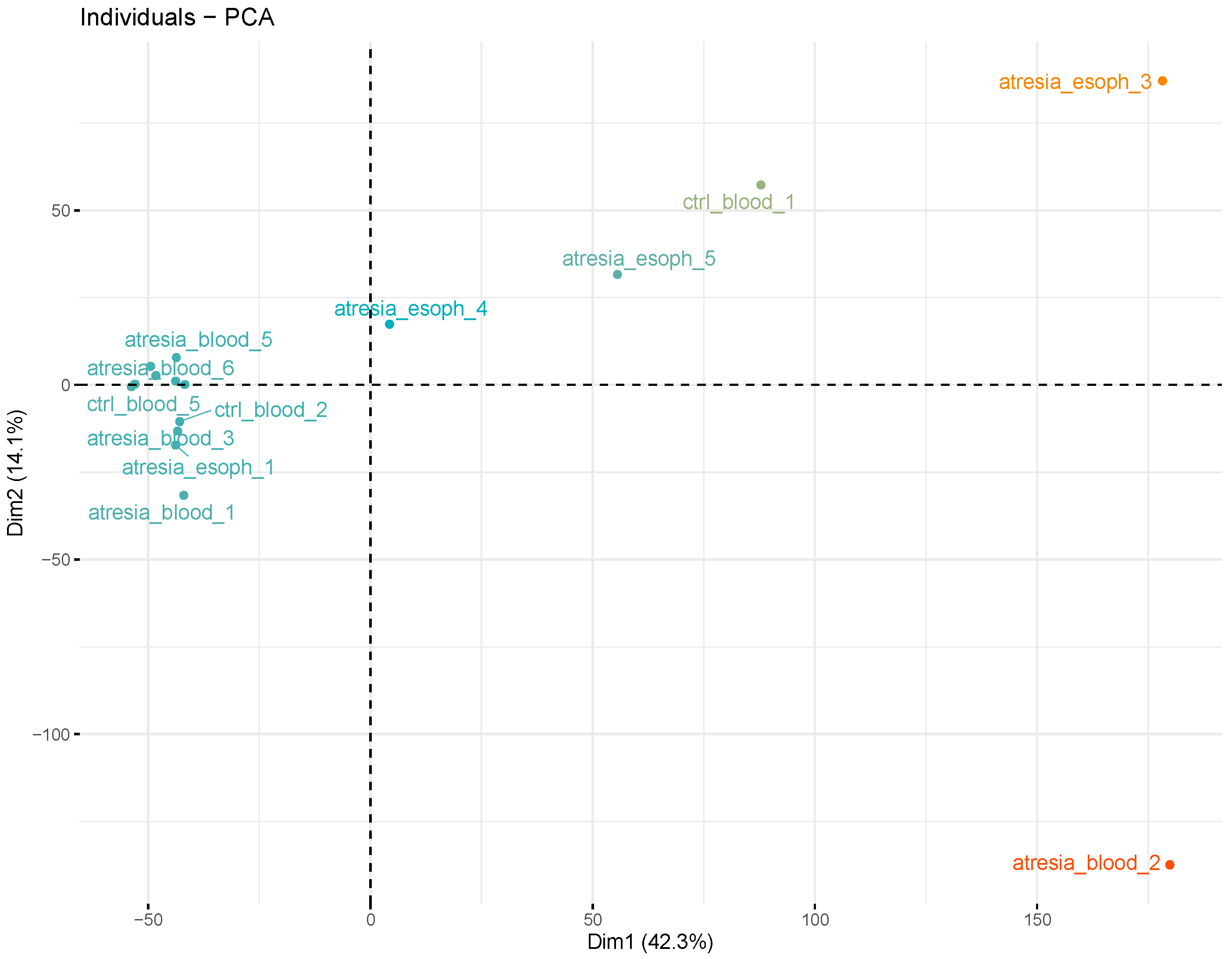
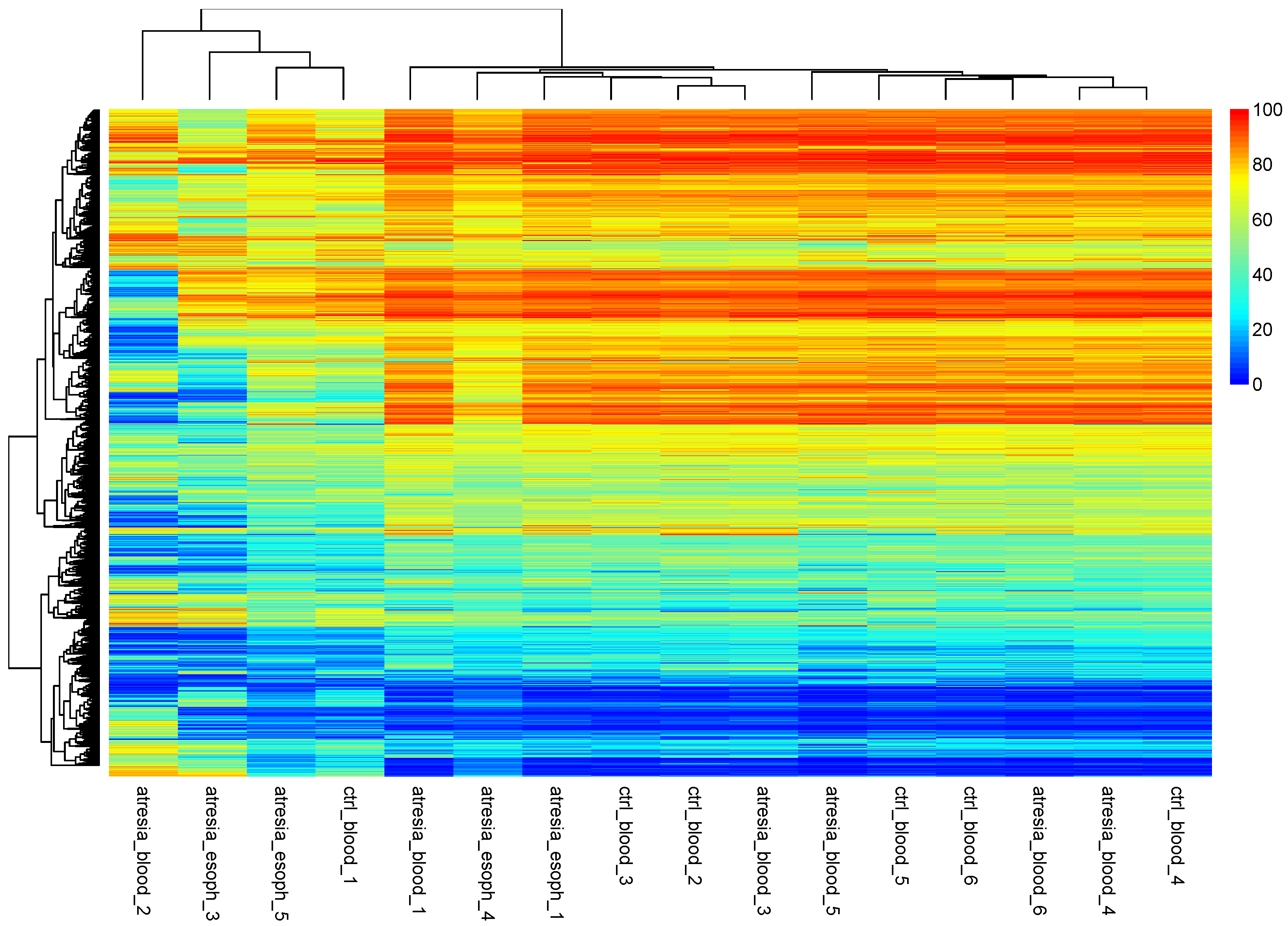
3.2. Unsupervised Analyses
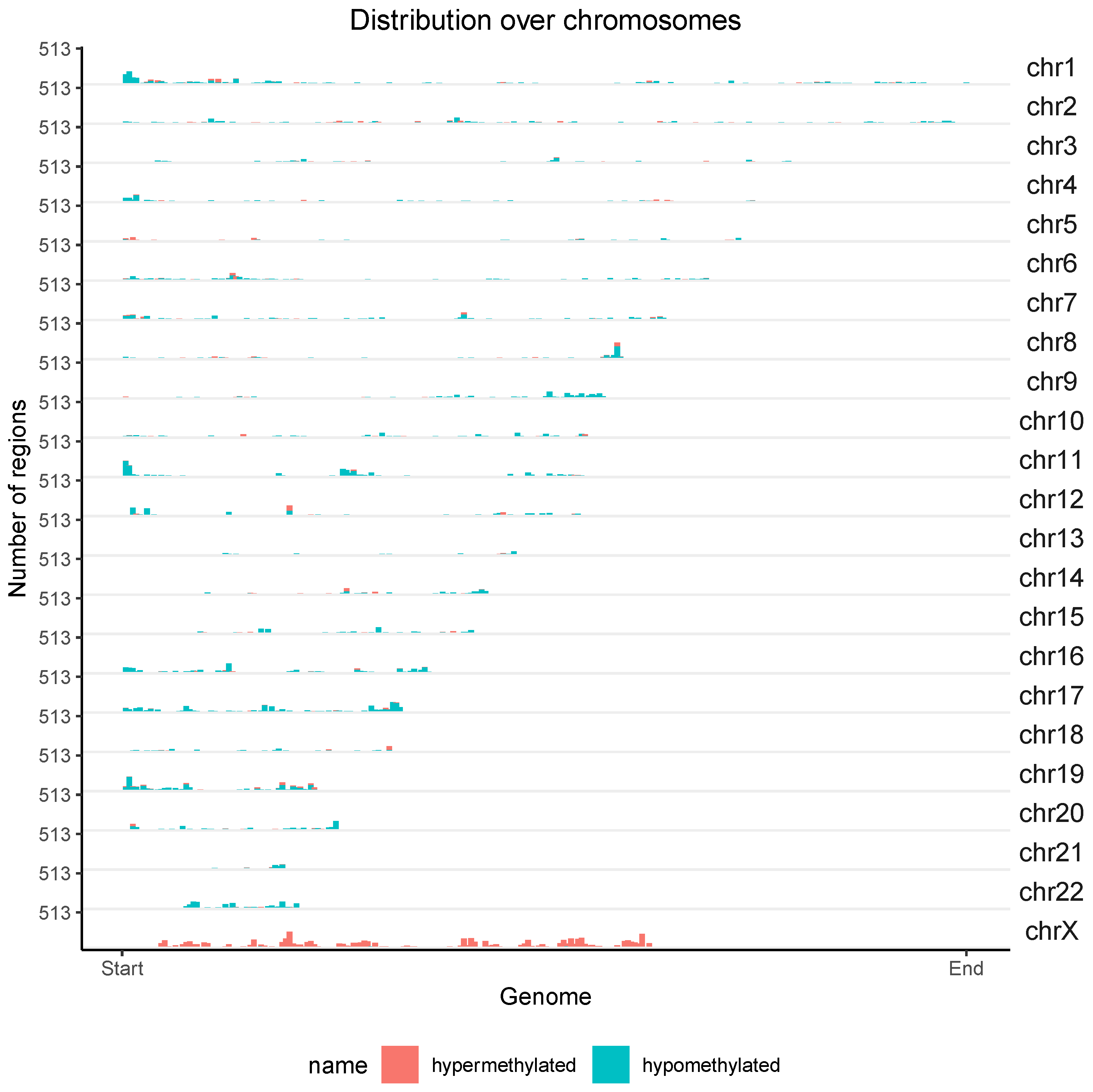
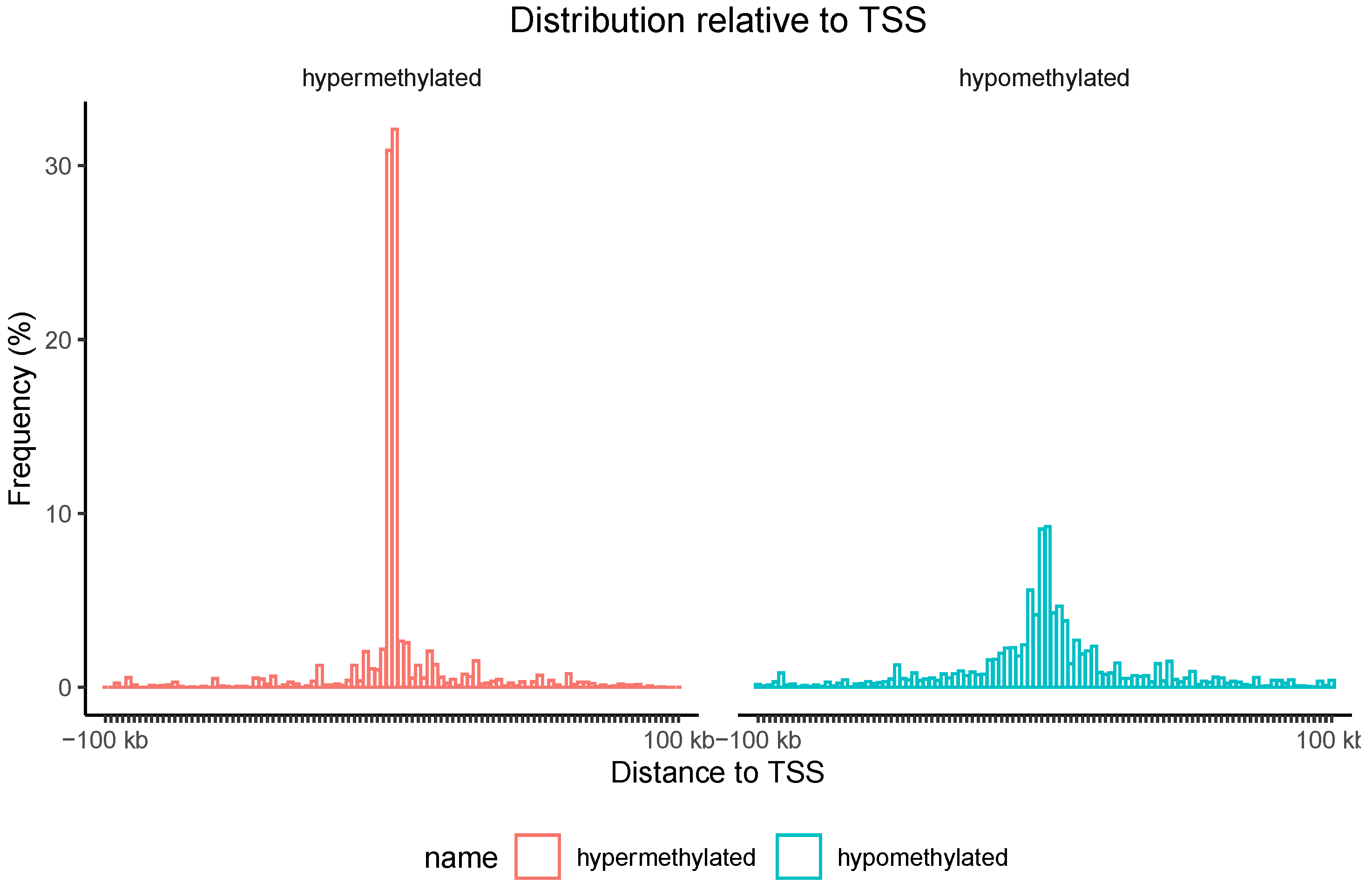
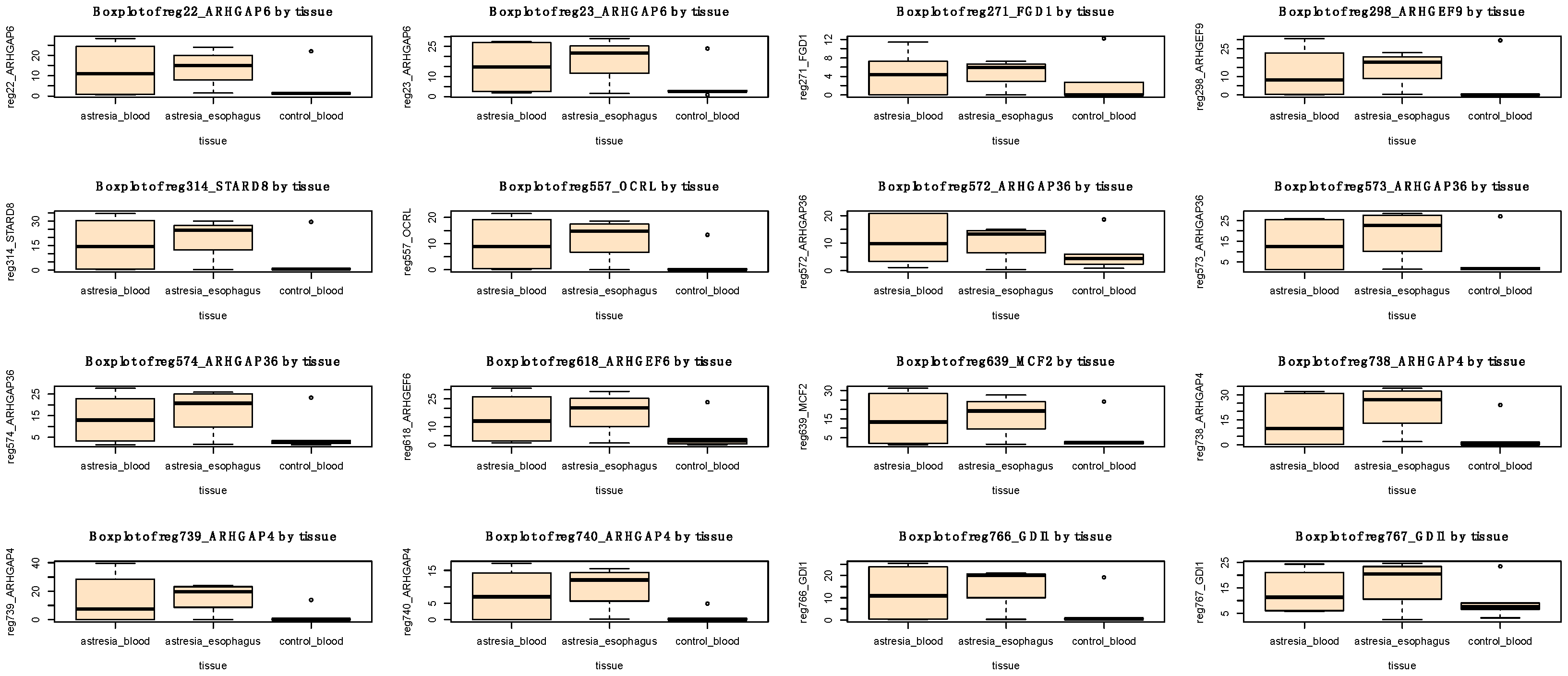
3.3. Differential Methylation (DM) Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pedersen, R.N.; Calzolari, E.; Husby, S.; Garne, E. Oesophageal atresia: Prevalence, prenatal diagnosis and associated anomalies in 23 European regions. Arch. Dis. Child. 2012, 97, 227–232. [Google Scholar] [CrossRef]
- Nassar, N.; Leoncini, E.; Amar, E.; Arteaga-Vázquez, J.; Bakker, M.K.; Bower, C.; Canfield, M.A.; Castilla, E.E.; Cocchi, G.; Correa, A.; et al. Prevalence of esophageal atresia among 18 international birth defects surveillance programs. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 893–899. [Google Scholar] [CrossRef]
- Gross, R. The Surgery of Infancy and Chilhood; WB Saunders: Philadelphia, PA, USA, 1953. [Google Scholar]
- Spitz, L. Oesophageal atresia. Orphanet J. Rare Dis. 2007, 2, 1–13. [Google Scholar] [CrossRef]
- Patkowski, D. Thoracoscopic approach for oesophageal atresia: A real game changer? J. Pediatr. Surg. 2023, 58, 204–208. [Google Scholar] [CrossRef]
- Brosens, E.; Brouwer, R.W.W.; Douben, H.; van Bever, Y.; Brooks, A.S.; Wijnen, R.M.H.; van Ijcken, W.F.J.; Tibboel, D.; Rottier, R.J.; de Klein, A. Heritability and De Novo Mutations in Oesophageal Atresia and Tracheoesophageal Fistula Aetiology. Genes 2021, 12, 1595. [Google Scholar] [CrossRef]
- Edwards, N.A.; Shacham-Silverberg, V.; Weitz, L.; Kingma, P.S.; Shen, Y.; Wells, J.M.; Chung, W.K.; Zorn, A.M. Developmental basis of trachea-esophageal birth defects. Dev. Biol. 2021, 477, 85–97. [Google Scholar] [CrossRef]
- Weissbach, T.; Kassif, E.; Kushnir, A.; Shust-Barequet, S.; Leibovitch, L.; Eliasi, E.; Elkan-Miller, T.; Zajicek, M.; Yinon, Y.; Weisz, B. Esophageal atresia in twins compared to singletons: In utero manifestation and characteristics. Prenat. Diagn. 2020, 40, 1418–1425. [Google Scholar] [CrossRef]
- Maroszyńska, I.; Fortecka-Piestrzeniewicz, K.; Niedźwiecka, M.; Żarkowska-Szaniawska, A. Isolated esophageal atresia in both premature twins. Pediatr. Polska 2015, 90, 91–93. [Google Scholar] [CrossRef]
- Klaniewska, M.; Toczewski, K.; Rozensztrauch, A.; Bloch, M.; Dzielendziak, A.; Gasperowicz, P.; Slezak, R.; Ploski, R.; Rydzanicz, M.; Smigiel, R.; et al. Occurrence of Esophageal Atresia With Tracheoesophageal Fistula in Siblings From Three-Generation Family Affected by Variable Expressivity MYCN Mutation: A Case Report. Front. Pediatr. 2021, 9, 783553. [Google Scholar] [CrossRef]
- Felson, J. What can we learn from twin studies? A comprehensive evaluation of the equal environments assumption. Soc. Sci. Res. 2014, 43, 184–199. [Google Scholar] [CrossRef]
- Ginzel, M.; Huber, N.; Bauer, L.; Kluth, D.; Metzger, R. Development of the foregut and the formation of the trachea and esophagus in rat embryos. A symphony of confusion. Front. Cell Dev. Biol. 2023, 11, 1092753. [Google Scholar] [CrossRef] [PubMed]
- Billmyre, K.K.; Hutson, M.; Klingensmith, J. One shall become two: Separation of the esophagus and trachea from the common foregut tube. Dev. Dyn. 2014, 244, 277–288. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.M.; Felix, J.F.; de Klein, A.; Tibboel, D. Etiology of Esophageal Atresia and Tracheoesophageal Fistula: “Mind the Gap”. Curr. Gastroenterol. Rep. 2010, 12, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef]
- Wu, H.; Caffo, B.; Jaffee, H.A.; Irizarry, R.A.; Feinberg, A.P. Redefining CpG islands using hidden Markov models. Biostatistics 2010, 11, 499–514. [Google Scholar] [CrossRef]
- Löffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Moarii, M.; Boeva, V.; Vert, J.-P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 873. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Koo, B.; Yang, J.; Kim, S. Metheor: Ultrafast DNA methylation heterogeneity calculation from bisulfite read alignments. PLoS Comput. Biol. 2023, 19, e1010946. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Nebel, A.; Franke, A.; Walter, J.; Lengauer, T.; Bock, C.; Müller, F.; List, M. Quantitative comparison of within-sample heterogeneity scores for DNA methylation data. Nucleic Acids Res. 2020, 48, e46. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Xue, H.; Wei, L.; Jin, J.; Tian, X.; Wang, Q. High heterogeneity undermines generalization of differential expression results in RNA-Seq analysis. Hum. Genom. 2021, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sears, R.L.; Xing, X.; Zhang, B.; Li, D.; Rockweiler, N.B.; Jang, H.S.; Choudhary, M.N.; Lee, H.J.; Lowdon, R.F.; et al. Tissue-specific DNA methylation is conserved across human, mouse, and rat, and driven by primary sequence conservation. BMC Genom. 2017, 18, 724. [Google Scholar] [CrossRef]
- Salas, L.A.; Koestler, D.C.; Butler, R.A.; Hansen, H.M.; Wiencke, J.K.; Kelsey, K.T.; Christensen, B.C. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018, 19, 64. [Google Scholar] [CrossRef]
- Sadok, A.; Marshall, C.J. Rho GTPases. Small GTPases 2014, 5, e983878. [Google Scholar] [CrossRef]
- Van Aelst, L.; Symons, M. Role of Rho family GTPases in epithelial morphogenesis. Genes Dev. 2002, 16, 1032–1054. [Google Scholar] [CrossRef]
- Rodríguez-Fdez, S.; Bustelo, X.R. Rho GTPases in Skeletal Muscle Development and Homeostasis. Cells 2021, 10, 2984. [Google Scholar] [CrossRef]
- Dahmene, M.; Quirion, L.; Laurin, M. High Throughput strategies Aimed at Closing the GAP in Our Knowledge of Rho GTPase Signaling. Cells 2020, 9, 1430. [Google Scholar] [CrossRef]
- El Masri, R.; Delon, J. RHO GTPases: From new partners to complex immune syndromes. Nat. Rev. Immunol. 2021, 21, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ji, H.; Xu, Q.; Zhang, X.; Cao, X.; Chen, Y.; Shao, M.; Wu, Z.; Zhang, J.; Lu, C.; et al. Congenital biliary atresia is correlated with disrupted cell junctions and polarity caused by Cdc42 in-sufficiency in the liver. Theranostics 2021, 11, 7262–7275. [Google Scholar] [CrossRef] [PubMed]
- Depaepe, A.; Dolk, H.; Lechat, M.F. The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch. Dis. Child. 1993, 68, 743–748. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a | 1b | 2a | 2b | 3a | 3b | 4a | 4b | 5a | 5b | 6a | 6b | |
| Zygostity | Dizygotic | Monozygotic | Dizygotic | Monozygotic | Monozygotic | Dizygotic | ||||||
| Gender | F | F | F | F | F | M | M | M | M | M | M | M |
| EA Gross type | C (t) | C | C (t) | C (t) | C (t) | C | ||||||
| Symbol | Gene Name | OMIM |
|---|---|---|
| ARHGAP36 | Rho GTPase activating protein 36 | 300937 |
| ARHGAP4 | Rho GTPase activating protein 4 | 300023 |
| ARHGAP6 | Rho GTPase activating protein 6 | 300118 |
| ARHGEF6 | Rac/Cdc42 guanine nucleotide exchange factor 6 | 300267 |
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor 9 | 300429 |
| FGD1 | FYVE, RhoGEF, and PH domain containing 1 | 300546 |
| GDI1 | GDP dissociation inhibitor 1 | 300104 |
| MCF2 | MCF.2-cell-line-derived transforming sequence | 311030 |
| OCRL | OCRL inositol polyphosphate-5-phosphatase | 300535 |
| STARD8 | StAR-related lipid transfer domain containing 8 | 300689 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Błoch, M.; Gasperowicz, P.; Gerus, S.; Rasiewicz, K.; Lebioda, A.; Skiba, P.; Płoski, R.; Patkowski, D.; Karpiński, P.; Śmigiel, R. Epigenetic Findings in Twins with Esophageal Atresia. Genes 2023, 14, 1822. https://doi.org/10.3390/genes14091822
Błoch M, Gasperowicz P, Gerus S, Rasiewicz K, Lebioda A, Skiba P, Płoski R, Patkowski D, Karpiński P, Śmigiel R. Epigenetic Findings in Twins with Esophageal Atresia. Genes. 2023; 14(9):1822. https://doi.org/10.3390/genes14091822
Chicago/Turabian StyleBłoch, Michal, Piotr Gasperowicz, Sylwester Gerus, Katarzyna Rasiewicz, Arleta Lebioda, Pawel Skiba, Rafal Płoski, Dariusz Patkowski, Pawel Karpiński, and Robert Śmigiel. 2023. "Epigenetic Findings in Twins with Esophageal Atresia" Genes 14, no. 9: 1822. https://doi.org/10.3390/genes14091822
APA StyleBłoch, M., Gasperowicz, P., Gerus, S., Rasiewicz, K., Lebioda, A., Skiba, P., Płoski, R., Patkowski, D., Karpiński, P., & Śmigiel, R. (2023). Epigenetic Findings in Twins with Esophageal Atresia. Genes, 14(9), 1822. https://doi.org/10.3390/genes14091822