
Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis of Putative Binding Sites (E-Boxes) in the Human CHRDL1 Gene Upstream Sequence
2.2. Cell Culture
2.3. Plasmid Constructs and Site-Directed Mutagenesis
2.4. In Vitro Protein Synthesis, and Electrophoretic Mobility Shift Assay (EMSA)
2.5. Luciferase Reporter Gene Assay
3. Results
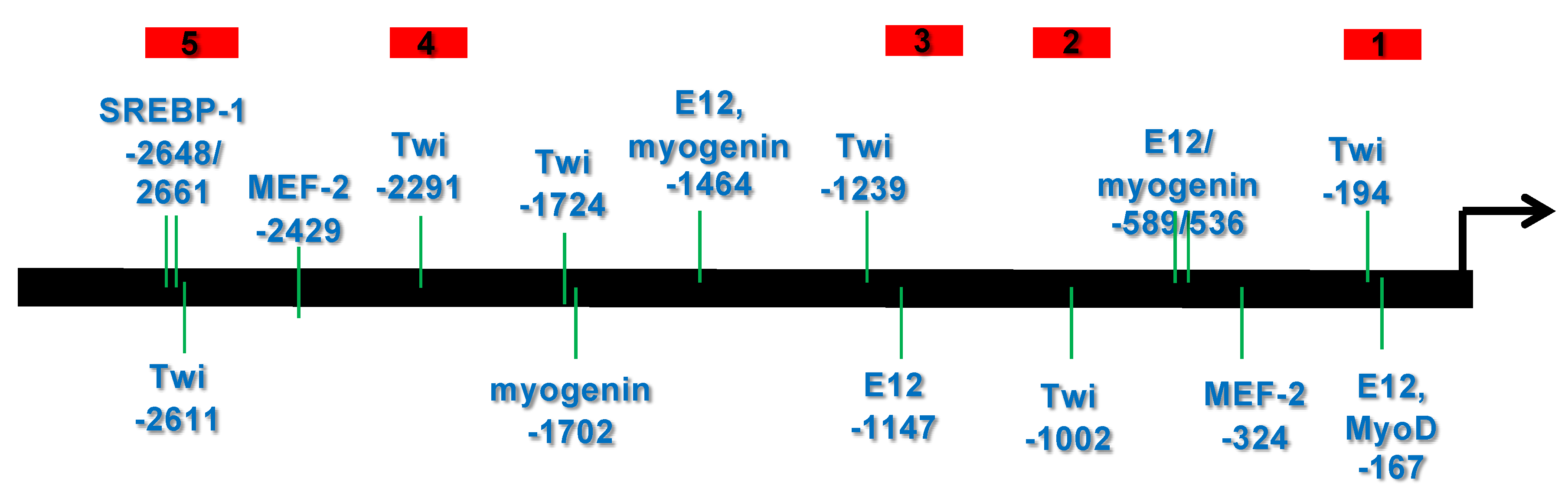
3.1. Bioinformatic Analysis Reveals That the 5′ Upstream Region of the Human CHRDL1 Gene Contains Putative Transcription Factor Binding Sites Which Are Conserved in Mammals
3.1.1. Transcription Element Search System (TESS) Analysis
3.1.2. mVISTA Analysis of Mammalian Chordin-like 1 Genes
3.2. Several bHLH Transcription Factors Have DNA Binding Activity to Upstream Region Sequence of the Human CHRDL1 Gene
3.3. SREBP1c/Binds to the Most Upstream Ebox Site along TWIST1 and Wild Type TWIST2
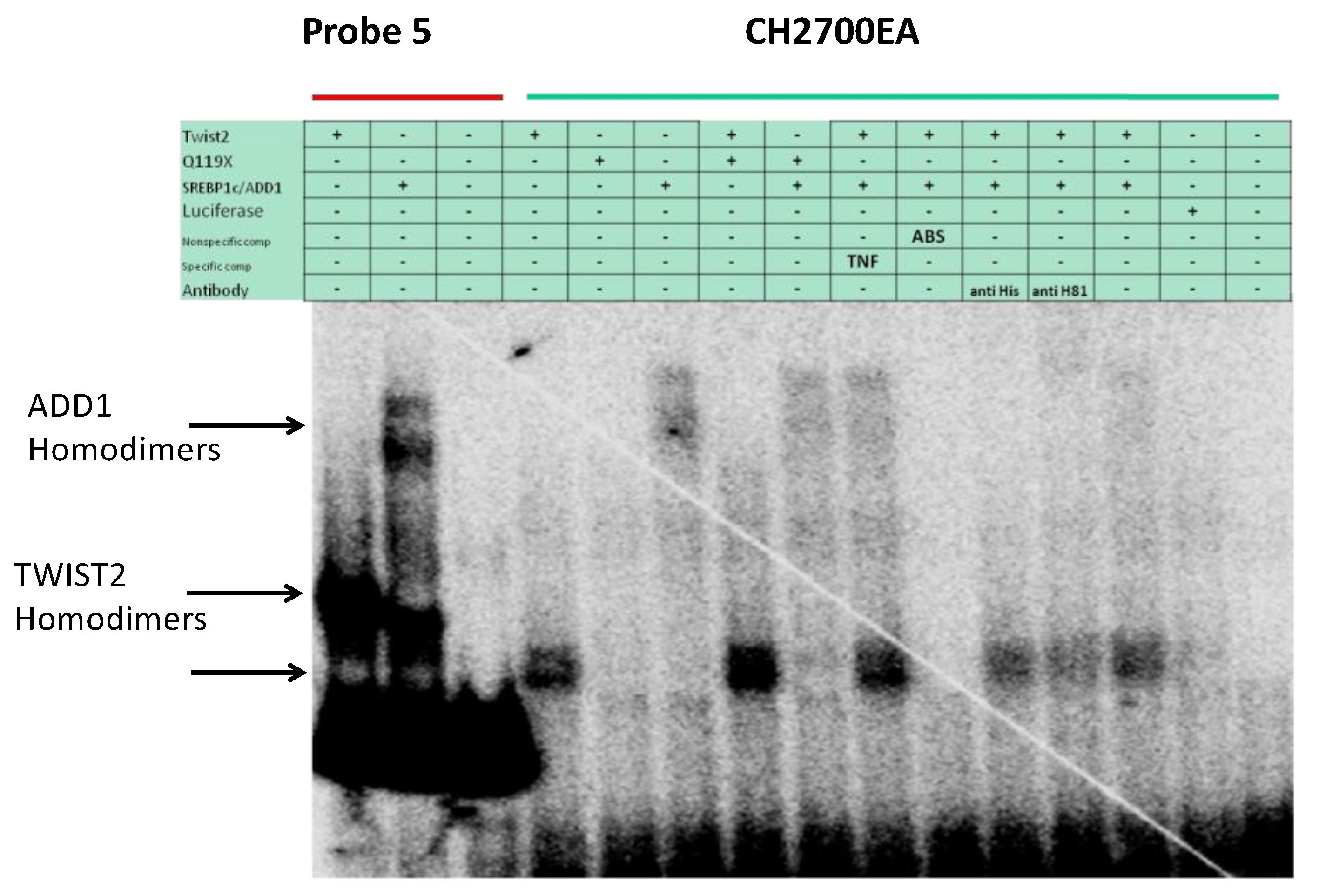
3.4. SREBP1c Can Bind Both the CHRDL1 Gene −2661 and −2648 E-Boxes While TWIST2 Prefers to Bind the −2648 E-Box, SREBP1c and TWIST2 Compete for Binding to These E-Boxes
3.5. The TWIST2 Q119X Mutant Protein Can Bind the Most Upstream Site of the CHRDL1 Gene as a Heterodimer with E12
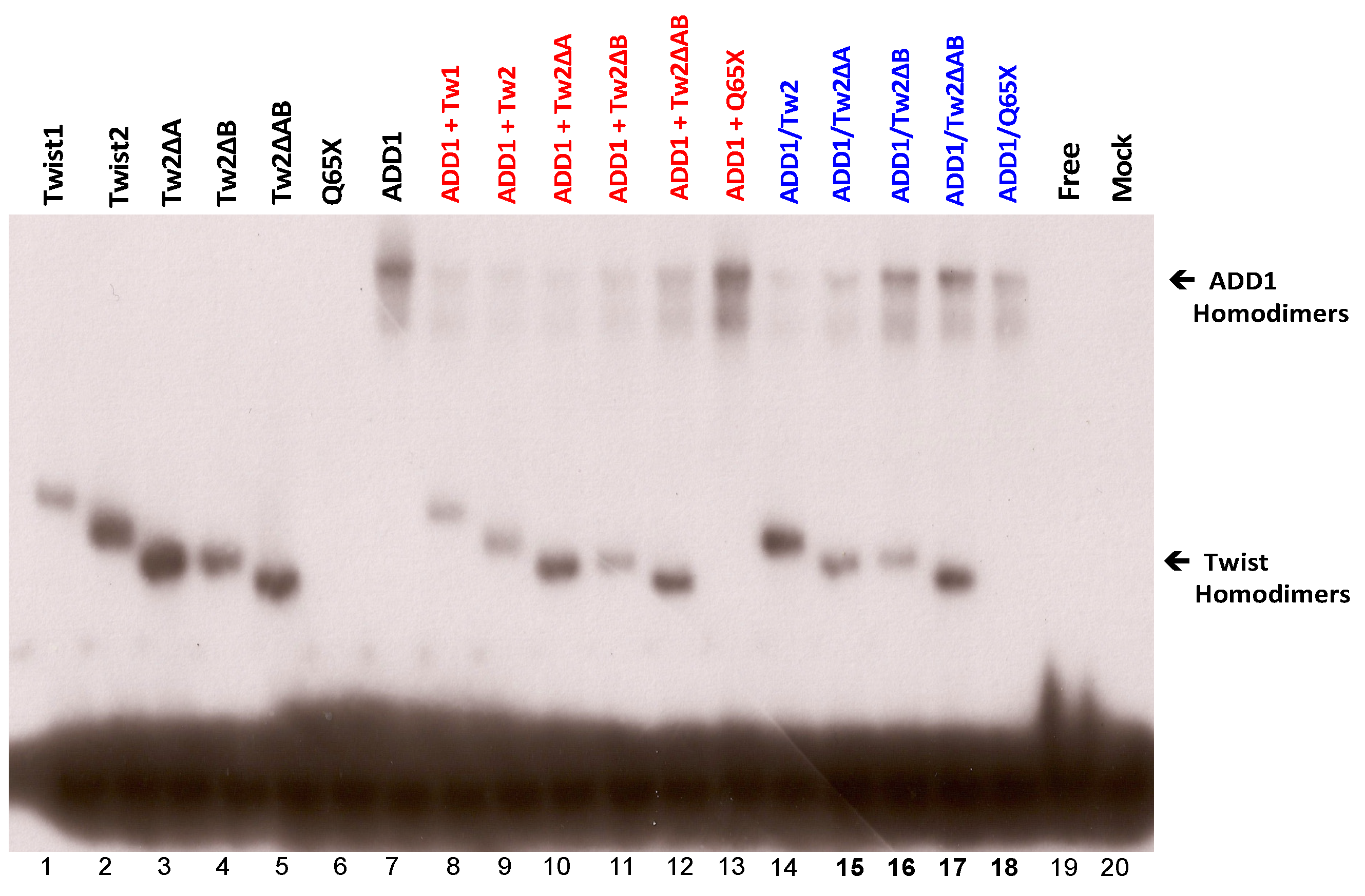
3.6. TWIST2 Uses Its Second Conserved Sub-Motif (SEEE) to Regulate the DNA-Binding Activity of SREBP1c
3.7. The Glycine-Rich Regions Present in TWIST1 Influence Its Interaction with SREBP1c
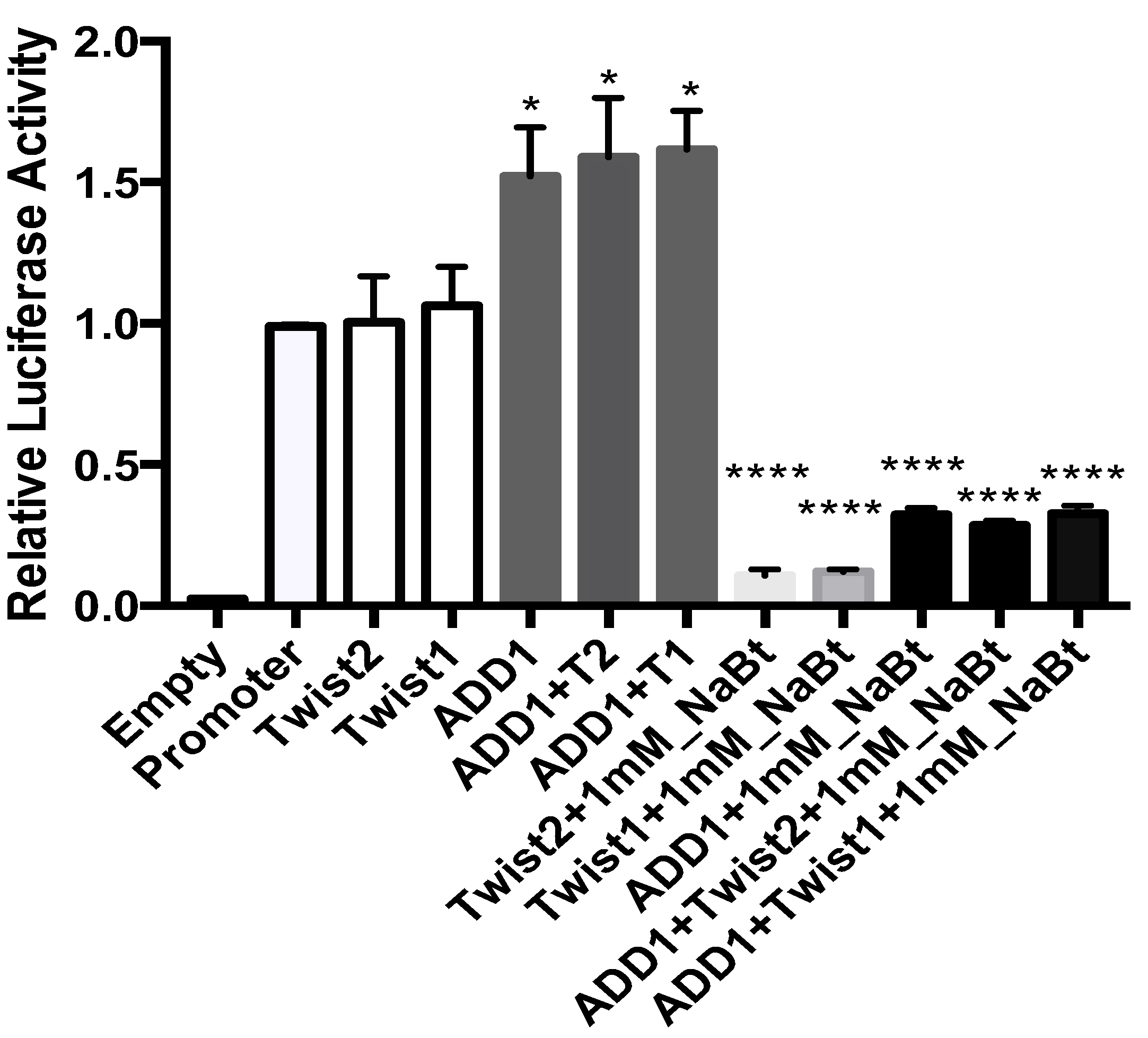
3.8. SREBP1c and the TWIST2 Q119X Mutant Protein Activate the Luciferase Reporter but Wild Type TWIST2 Blocks SREBP1c-Mediated Activation
3.9. In HeLa Cells, TWIST2 and Its N-Terminal Mutant Forms Reduce the Transactivating Activity of SREBP1c
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Setleis, H.; Kramer, B.; Valcarcel, M.; Einhorn, A.H. Congenital ectodermal dysplasia of the face. Pediatrics 1963, 32, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.D.; Golabi, M.; Lacassie, Y.; Hall, B.; Seto, S. Expanded phenotype and ethnicity in Setleis syndrome. Am. J. Med. Genet. 1989, 34, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Artlich, A.; Schwinger, E.; Meinecke, P. Setleis (bitemporal ‘forceps marks’) syndrome in a German family: Evidence for autosomal dominant inheritance. Clin. Dysmorphol. 1992, 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Kuno, T.; Hamasaki, Y.; Miyazaki, S.; Miyabara, S.; Narisawa, Y. Setleis bitemporal “Forceps Marks” Syndrome and its pathogenesis: A case report. Acta Paediatr. Jpn. 1991, 33, 186–190. [Google Scholar] [CrossRef]
- McGaughran, J.; Aftimos, S. Setleis syndrome: Three new cases and a review of the literature. Am. J. Med. Genet. 2002, 111, 376–380. [Google Scholar] [CrossRef] [PubMed]
- al-Gazali, L.I.; al-Talabani, J. Setleis syndrome: Autosomal recessive or autosomal dominant inheritance? Clin. Dysmorphol. 1996, 5, 249–253. [Google Scholar] [CrossRef]
- Tukel, T.; Šošić, D.; Al-Gazali, L.I.; Erazo, M.; Casasnovas, J.; Franco, H.L.; Richardson, J.A.; Olson, E.N.; Cadilla, C.L.; Desnick, R.J. Homozygous Nonsense Mutations in TWIST2 Cause Setleis Syndrome. Am. J. Hum. Genet. 2010, 87, 289–296. [Google Scholar] [CrossRef]
- Li, L.; Cserjesi, P.; Olson, E.N. Dermo-1: A novel twist-related bHLH protein expressed in the developing dermis. Dev. Biol. 1995, 172, 280–292. [Google Scholar] [CrossRef]
- Bialek, P.; Kern, B.; Yang, X.; Schrock, M.; Šošić, D.; Hong, N.; Wu, H.; Yu, K.; Ornitz, D.M.; Olson, E.N.; et al. A twist code determines the onset of osteoblast differentiation. Dev. Cell 2004, 6, 423–435. [Google Scholar] [CrossRef]
- Cervantes-Barragán, D.E.; Villarroel, C.E.; Medrano-Hernández, A.; Durán-McKinster, C.; Bosch-Canto, V.; Del-Castillo, V.; Nazarenko, I.; Yang, A.; Desnick, R.J. Setleis syndrome in Mexican-Nahua sibs due to a homozygous TWIST2 frameshift mutation and partial expression in heterozygotes: Review of the focal facial dermal dysplasias and subtype reclassification. J. Med. Genet. 2011, 48, 716–720. [Google Scholar] [CrossRef]
- Rosti, R.O.; Uyguner, Z.O.; Nazarenko, I.; Bekerecioglu, M.; Cadilla, C.L.; Ozgur, H.; Lee, B.H.; Aggarwal, A.K.; Pehlivan, S.; Desnick, R.J. Setleis syndrome: Clinical, molecular and structural studies of the first TWIST2 missense mutation. Clin. Genet. 2015, 88, 489–493. [Google Scholar] [CrossRef]
- Girisha, K.M.; Bidchol, A.M.; Sarpangala, M.K.; Satyamoorthy, K. A novel frameshift mutation in TWIST2 gene causing Setleis syndrome. Indian J. Pediatr. 2014, 81, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.D.; Norby, A.R.; Rosenfeld, J.A.; Proud, V.K.; Spangler, B.E.; Ming, J.E.; Chisholm, E.; Zackai, E.H.; Lee, B.H.; Edelmann, L.; et al. Chromosome 1p36.22p36.21 duplications/triplication causes Setleis syndrome (focal facial dermal dysplasia type III). Am. J. Med. Genet. A 2015, 167A, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kasparis, C.; Chen, B.; Mei, H.; Edelmann, L.; Moss, C.; Weaver, D.D.; Desnick, R.J. Setleis syndrome due to inheritance of the 1p36.22p36.21 duplication: Evidence for lack of penetrance. J. Hum. Genet. 2015, 60, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Marchegiani, S.; Davis, T.; Tessadori, F.; van Haaften, G.; Brancati, F.; Hoischen, A.; Huang, H.; Valkanas, E.; Pusey, B.; Schanze, D.; et al. Recurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes. Am. J. Hum. Genet. 2015, 97, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Šošić, D.; Richardson, J.A.; Yu, K.; Ornitz, D.M.; Olson, E.N. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 2003, 112, 169–180. [Google Scholar] [CrossRef]
- Lee, M.S.; Lowe, G.; Flanagan, S.; Kuchler, K.; Glackin, C.A. Human dermo-1 has attributes similar to twist in early bone development. Bone 2000, 27, 591–602. [Google Scholar] [CrossRef]
- el Ghouzzi, V.; Le Merrer, M.; Perrin-Schmitt, F.; Lajeunie, E.; Benit, P.; Renier, D.; Bourgeois, P.; Bolcato-Bellemin, A.L.; Munnich, A.; Bonaventure, J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat. Genet. 1997, 15, 42–46. [Google Scholar] [CrossRef]
- Kunz, J.; Hudler, M.; Fritz, B. Identification of a frameshift mutation in the gene TWIST in a family affected with Robinow-Sorauf syndrome. J. Med. Genet. 1999, 36, 650–652. [Google Scholar] [CrossRef]
- Kim, S.; Twigg, S.R.F.; Scanlon, V.A.; Chandra, A.; Hansen, T.J.; Alsubait, A.; Fenwick, A.L.; McGowan, S.J.; Lord, H.; Lester, T.; et al. Localized TWIST1 and TWIST2 basic domain substitutions cause four distinct human diseases that can be modeled in Caenorhabditis elegans. Hum. Mol. Genet. 2017, 26, 2118–2132. [Google Scholar] [CrossRef]
- Seto, M.L.; Hing, A.V.; Chang, J.; Hu, M.; Kapp-Simon, K.A.; Patel, P.K.; Burton, B.K.; Kane, A.A.; Smyth, M.D.; Hopper, R.; et al. Isolated sagittal and coronal craniosynostosis associated with TWIST box mutations. Am. J. Med. Genet. 2007, 143A, 678–686. [Google Scholar] [CrossRef]
- Howard, T.D.; Paznekas, W.A.; Green, E.D.; Chiang, L.C.; Ma, N.; Ortiz de Luna, R.I.; Garcia Delgado, C.; Gonzalez-Ramos, M.; Kline, A.D.; Jabs, E.W. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 1997, 15, 36–41. [Google Scholar] [CrossRef]
- Takenouchi, T.; Sakamoto, Y.; Sato, H.; Suzuki, H.; Uehara, T.; Ohsone, Y.; Kosaki, K. Ablepharon and craniosynostosis in a patient with a localized TWIST1 basic domain substitution. Am. J. Med. Genet. A 2018, 176, 2777–2780. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Fujimoto, M.; Lopes, A.; Wang, Y.X. Twist1 is a PPAR -inducible, negative-feedback regulator of PGC-1 in brown fat metabolism. Cell 2009, 137, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, A.T.; Laurencikiene, J.; Mejhert, N.; Naslund, E.; Bouloumie, A.; Dahlman, I.; Arner, P.; Ryden, M. A possible inflammatory role of Twist1 in human white adipocytes. Diabetes 2010, 59, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, H.H.; Park, J.; Yoo, E.J.; Glackin, C.A.; Choi, Y.I.; Jeon, S.H.; Seong, R.H.; Park, S.D.; Kim, J.B. Twist2, a novel ADD1/SREBP1c interacting protein, represses the transcriptional activity of ADD1/SREBP1c. Nucleic Acids Res. 2003, 31, 7165–7174. [Google Scholar] [CrossRef] [PubMed]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or Separate Factors? Roles of Twist1 and Twist2 as molecular switches in gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef]
- Franco, H.L.; Casasnovas, J.J.; Leon, R.G.; Friesel, R.; Ge, Y.; Desnick, R.J.; Cadilla, C.L. Nonsense mutations of the bHLH transcription factor TWIST2 found in Setleis Syndrome patients cause dysregulation of periostin. Int. J. Biochem. Cell Biol. 2011, 43, 1523–1531. [Google Scholar] [CrossRef]
- Crespo, N.E.; Torres-Bracero, A.; Renta, J.Y.; Desnick, R.J.; Cadilla, C.L. Expression Profiling Identifies TWIST2 Target Genes in Setleis Syndrome Patient Fibroblast and Lymphoblast Cells. Int. J. Environ. Res. Public Health 2021, 18, 1997. [Google Scholar] [CrossRef]
- Coffinier, C.; Tran, U.; Larraín, J.; De Robertis, E.M. Neuralin-1 is a novel Chordin-related molecule expressed in the mouse neural plate. Mech. Dev. 2001, 100, 119–122. [Google Scholar] [CrossRef]
- Sakuta, H.; Suzuki, R.; Takahashi, H.; Kato, A.; Shintani, T.; Iemura, S.; Yamamoto, T.; Ueno, N.; Noda, M. Ventroptin: A BMP-4 Antagonist Expressed in a Double-Gradient Pattern in the Retina. Science 2001, 293, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Nishimatsu, S.; Thomsen, G.H. Ventral mesoderm induction and patterning by bone morphogenetic protein heterodimers in Xenopus embryos. Mech. Dev. 1998, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Graf, D.; Malik, Z.; Hayano, S.; Mishina, Y. Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev. 2016, 27, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Lander, A.D.; Nie, Q. Computational analysis of BMP gradients in dorsal-ventral patterning of the zebrafish embryo. J. Theor. Biol. 2007, 248, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Matarin, M.; Gardner, J.C.; Kelberman, D.; Hassan, H.; Ang, W.; Michaelides, M.; Ruddle, J.B.; Pennell, C.E.; Yazar, S.; et al. X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am. J. Hum. Genet. 2012, 90, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.E.; Cheong, S.S.; Hysi, P.G.; Venturini, C.; Plagnol, V.; Ruddle, J.B.; Ali, H.; Carnt, N.; Gardner, J.C.; Hassan, H.; et al. Association of CHRDL1 mutations and variants with X-linked megalocornea, Neuhäuser syndrome and central corneal thickness. PLoS ONE 2014, 9, e104163. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, Y.; Shu, G.; Chen, C.; Sullivan, D.A.; Kam, W.R.; Hann, S.; Fowler, M.; Warman, M.L. Ocular Manifestations of Chordin-like 1 Knockout Mice. Cornea 2020, 39, 1145–1150. [Google Scholar] [CrossRef]
- Weaving, L.; Mihelec, M.; Storen, R.; Šošić, D.; Grigg, J.R.; Tam, P.P.; Jamieson, R.V. Twist2: Role in corneal stromal keratocyte proliferation and corneal thickness. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5561–5570. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Toth, J.I.; Datta, S.; Athanikar, J.N.; Freedman, L.P.; Osborne, T.F. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol. Cell Biol. 2004, 24, 8288–8300. [Google Scholar] [CrossRef]
- Linser, R.; Salvi, N.; Briones, R.; Rovó, P.; de Groot, B.L.; Wagner, G. The membrane anchor of the transcriptional activator SREBP is characterized by intrinsic conformational flexibility. Proc. Natl. Acad. Sci. USA 2015, 112, 12390–12395. [Google Scholar] [CrossRef]
- Sun, H.; Wang, S.; Yang, Z.; Tian, L.; Li, X.; Zhou, J.; Wang, B. Chordin Like-1 Regulates Osteoblast and Adipocyte Differentiation Through Stabilizing Insulin-Like Growth Factor Binding Protein 3. Stem Cells 2023, 41, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Gonzalez-Mendez, R.R.; Cadilla, C.L. Evolution of the Twist Subfamily Vertebrate Proteins: Discovery of a Signature Motif and Origin of the Twist1 Glycine-Rich Motifs in the Amino-Terminus Disordered Domain. PLoS ONE 2016, 11, e0161029. [Google Scholar]
- Shug, J. Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence. Curr. Protoc. Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Pollakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Pollakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.; Dubchak, I. VISTA: Visualizing Global DNA Sequence Aligments of Arbitrary Length. Bioinformatics 2000, 16, 1046. [Google Scholar] [CrossRef] [PubMed]
- Schagat, T.; Paguio, A.; Kopish, K. Normalizing Genetic Reporter Assays: Approaches and Considerations for Increasing Consistency and Statistical Significance. Cell Notes 2007, 17, 9–12. [Google Scholar]
- Hao, Q.; Hansen, J.B.; Petersen, R.K.; Hallenborg, P.; Jorgensen, C.; Cinti, S.; Larsen, P.J.; Steffensen, K.R.; Wang, H.; Collins, S.; et al. ADD1/SREBP1c activates the PGC1-α promoter in brown adipocytes. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2010, 1801, 421–429. [Google Scholar] [CrossRef]
- Piccinin, S.; Tonin, E.; Sessa, S.; Demontis, S.; Rossi, S.; Pecciarini, L.; Zanatta, L.; Pivetta, F.; Grizzo, A.; Sonego, M.; et al. A “Twist box” Code of p53 Inactivation: Twist box: p53 Interaction Promotes p53 Degradation. Cancer Cell 2012, 22, 404–415. [Google Scholar] [CrossRef]
- Bonilla-Claudio, M.; Wang, J.; Bai, Y.; Klysik, E.; Selever, J.; Martin, J.F. Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development 2012, 139, 709–719. [Google Scholar] [CrossRef]
- Connerney, J.; Andreeva, V.; Leshem, Y.; Muentener, C.; Mercado, M.A.; Spicer, D.B. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. 2006, 235, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.B.; Aldrich, M.; Šošić, D.; Olson, E.N.; Friedman, A.D.; Lee, S.H.; Chen, S.Y. Twist-2 controls myeloid lineage development and function. PLoS Biol. 2008, 6, e316. [Google Scholar] [CrossRef] [PubMed]
- Laursen, K.B.; Mielke, E.; Iannaccone, P.; Fuchtbauer, E.M. Mechanism of transcriptional activation by the proto-oncogene Twist1. J. Biol. Chem. 2007, 282, 34623–34633. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Luukko, K.; Kettunen, P. BMP signaling in craniofacial development. Int. J. Dev. Biol. 2006, 50, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Hendrix, J.; Johnson, D.; Owens, G.K. Smooth muscle α-actin gene requires two E-boxes for proper expression in vivo and is a target of class I basic helix-loop-helix proteins. Circ. Res. 2003, 92, 840–847. [Google Scholar] [CrossRef]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.-R.; Fu, X.-D.; et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef]
- Connerney, J.; Andreeva, V.; Leshem, Y.; Mercado, M.A.; Dowell, K.; Yang, X.; Lindner, V.; Friesel, R.E.; Spicer, D.B. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev. Biol. 2008, 318, 323–334. [Google Scholar] [CrossRef]
- Rider, C.C.; Mulloy, B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 2010, 429, 1–12. [Google Scholar] [CrossRef]
- Foppiano, S.; Hu, D.; Marcucio, R.S. Signaling by bone morphogenetic proteins directs formation of an ectodermal signaling center that regulates craniofacial development. Dev. Biol. 2007, 312, 103–114. [Google Scholar] [CrossRef]
- Gong, X.Q.; Li, L. Dermo-1, a multifunctional basic helix-loop-helix protein, represses MyoD transactivation via the HLH domain, MEF2 interaction, and chromatin deacetylation. J. Biol. Chem. 2002, 277, 12310–12317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hassan, M.Q.; Li, Z.Y.; Stein, J.L.; Lian, J.B.; Van Wijnen, A.J.; Stein, G.S. Intricate gene regulatory networks of helix-loop-helix (HLH) proteins support regulation of bone-tissue related genes during osteoblast differentiation. J. Cell Biochem. 2008, 105, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Spicer, D.; Rhee, J.; Cheung, W.; Lassar, A. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH twist. Science 1996, 272, 1476–1480. [Google Scholar] [CrossRef]
- Jiang, L.; Hindmarch, C.; Rogers, M.F.; Campbell, C.; Waterfall, C.; Coghill, J.; Mathieson, P.W.; Welsh, G.I. RNA sequencing analysis of human podocytes reveals glucocorticoid regulated gene networks targeting non-immune pathways. Sci. Rep. 2016, 6, 35671. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Riley, S.; Fraser, D.; Al-Assaf, S.; Ishimura, E.; Wolever, T.; Phillips, G.O.; Phillips, A.O. Butyrate modulates TGF-beta1 generation and function: Potential renal benefit for Acacia(sen) SUPERGUM (gum arabic)? Kidney Int. 2006, 69, 257–265. [Google Scholar] [CrossRef][Green Version]
- Huang, R.; Lang, E.R.; Otto, W.R.; Christ, B.; Patel, K. Molecular and cellular analysis of embryonic avian tongue development. Anat. Embryol. Berl. 2001, 204, 179–187. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Y.; Cavender, A.C.; Wang, S.; Mansukhani, A.; D’Souza, R.N. Molecular studies on the roles of Runx2 and Twist1 in regulating FGF signaling. Dev. Dyn. 2012, 241, 1708–1715. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Twist Protein | Interactor | Domain Required by Twist | Effect | Domain Targeted in Interactor | Cell Line Used | Source |
|---|---|---|---|---|---|---|
| Twist2 | Runx2 | Twist Box (Last 20 residues) | Inhibition of Runx2 DNA binding | Runt domain | Osteoblasts | [9] |
| Twist2 | MEF2 | C-terminus 121–160 aa | Inhibition of MEF2 transactiva-tion activity | Transactivation domain | C3H10T1/2 cells | [61] |
| Twist2 | MyoD | C-terminus | Repression of MyoD transactiva-tion activity | Basic and HLH | C3H10T1/2 cells | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casasnovas-Nieves, J.J.; Rodríguez, Y.; Franco, H.L.; Cadilla, C.L. Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors. Genes 2023, 14, 1733. https://doi.org/10.3390/genes14091733
Casasnovas-Nieves JJ, Rodríguez Y, Franco HL, Cadilla CL. Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors. Genes. 2023; 14(9):1733. https://doi.org/10.3390/genes14091733
Chicago/Turabian StyleCasasnovas-Nieves, José J., Yacidzohara Rodríguez, Hector L. Franco, and Carmen L. Cadilla. 2023. "Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors" Genes 14, no. 9: 1733. https://doi.org/10.3390/genes14091733
APA StyleCasasnovas-Nieves, J. J., Rodríguez, Y., Franco, H. L., & Cadilla, C. L. (2023). Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors. Genes, 14(9), 1733. https://doi.org/10.3390/genes14091733